Pépite | Modélisation d’interactions biomoléculaires
98
0
0
Texte intégral
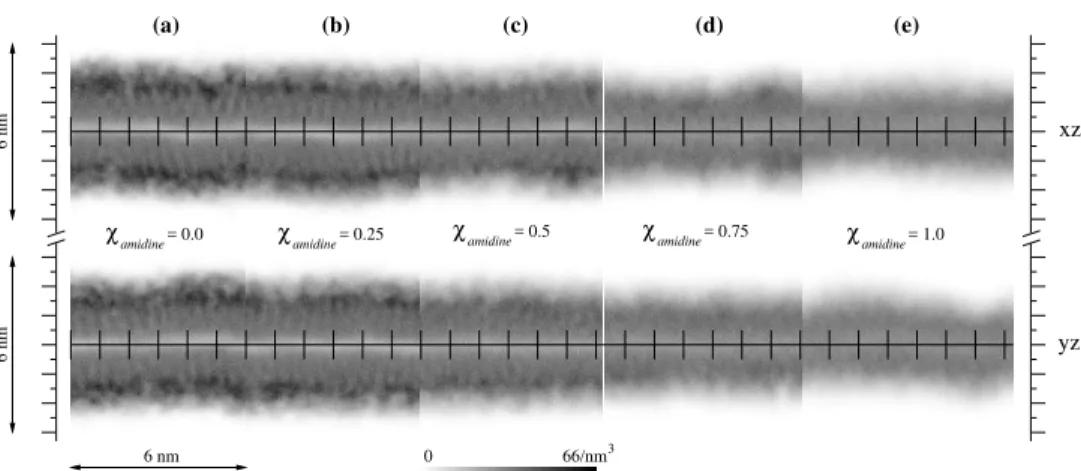
Figure

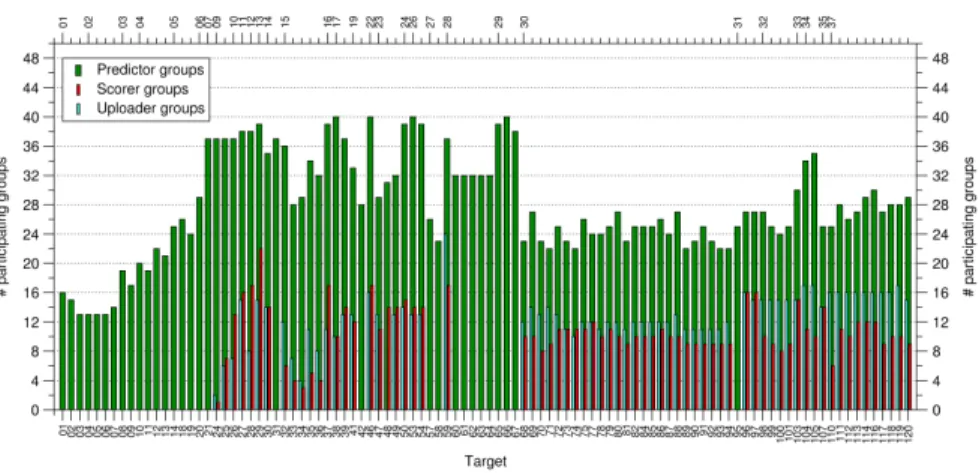


+7




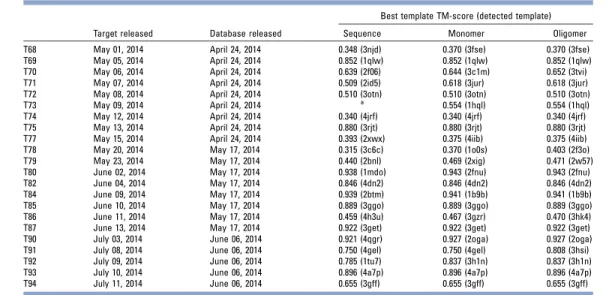
Documents relatifs
