Préparation de films à base de chitosane par voie fondue
Texte intégral
Figure
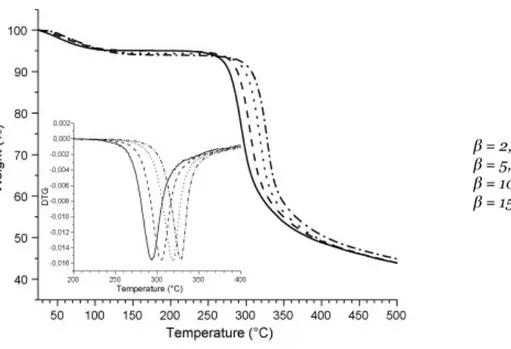
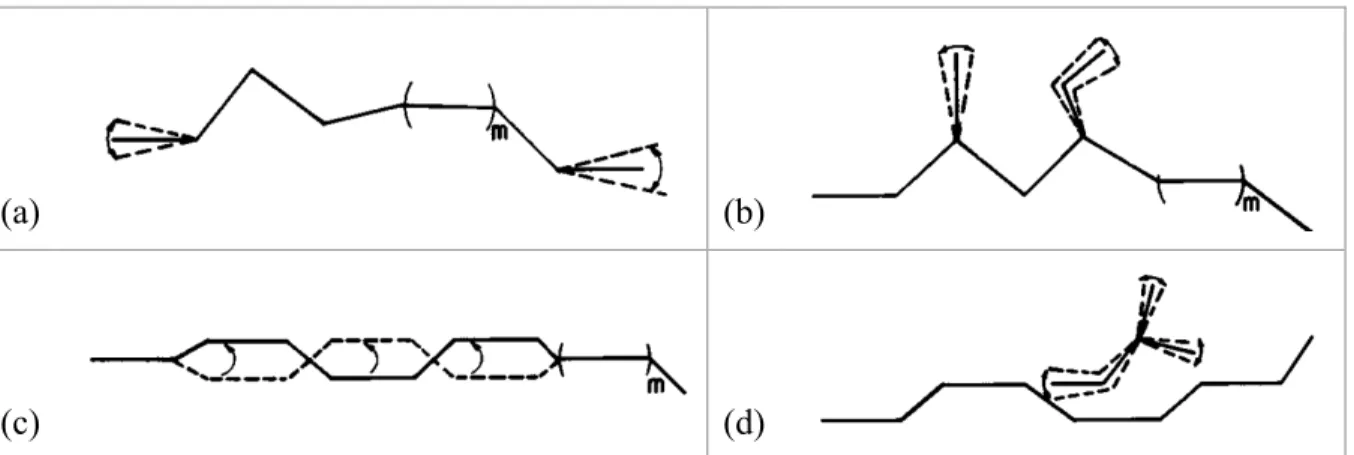
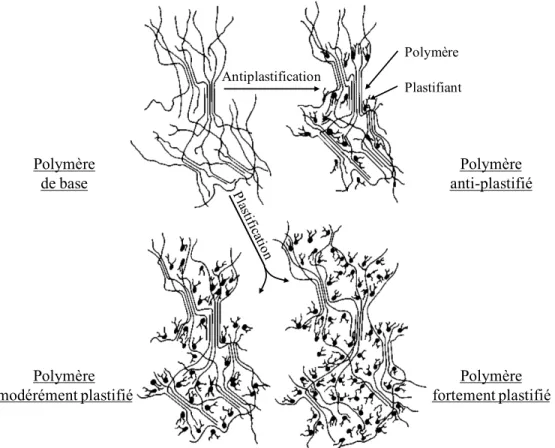
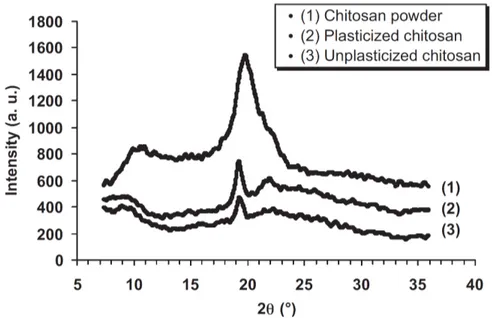
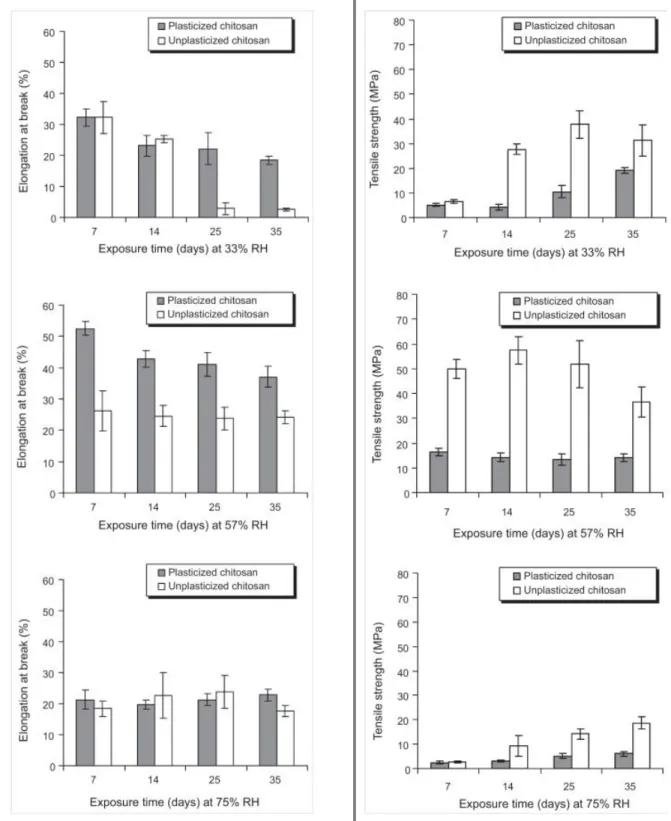
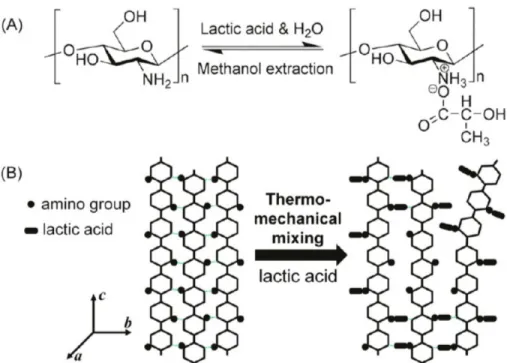
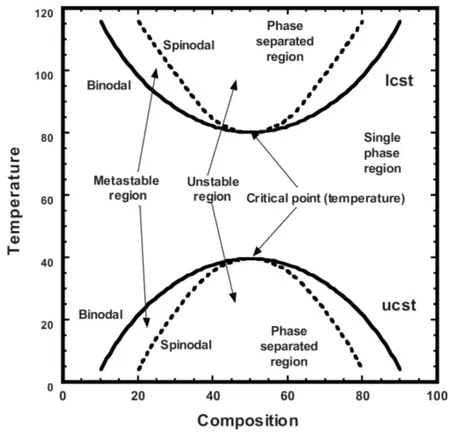
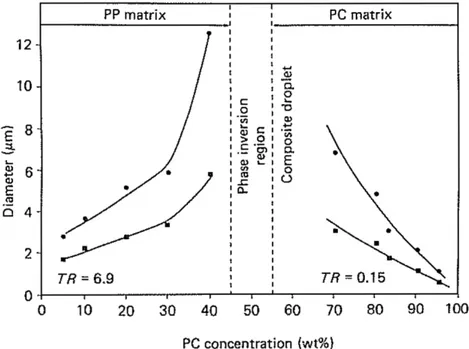
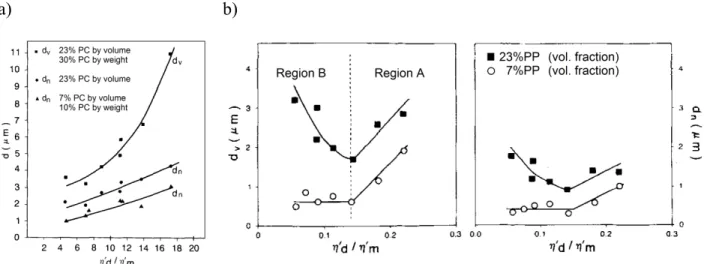
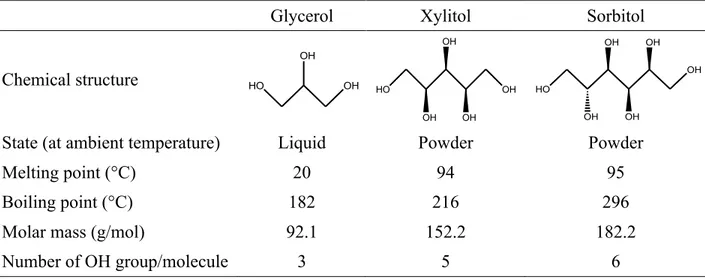
Documents relatifs
De plus, les couches minces à base de ZnO présentent des propriétés optiques et structurales de hautes qualités qui confirme l’exploitation de ces dispositifs pour des
En effet, la performance de ce chitosane comme agent floculant est plus faible que celle du polyacrylamide cationique commercial utilisé comme point de comparaison dans
Par ailleurs, les films élaborés à partir du latex de skim (fraction qui contient les petites particules et une plus grande proportion de non-caoutchoucs que la fraction 1)
Tableau V.11 : Coefficient Seebeck des films de Bi 2 Te 3 synthétisés par croissance électrochimique sur des pré-couches obtenues par cémentation pour différents temps. Les mesures
Ratios d’incorporation PM/VP en fonction de la vitesse de retrait dans les films minces trempés préparés à partir de solutions de PS-P4VP(41,5k- 17,5k) dans le
Sachant que le niveau d’ApoB en circulation a été réduit suite au traitement pour tous les groupes recevant des nanoparticules chitosane (92-10-5)/siARN-ApoB, il est important
En effet, des études antérieures ont démontré qu’après deux semaines, les implants chitosane-GP/sang solidifiés in situ promeuvent le recrutement d’ostéoclastes
ultrasonique en utilisant du méthanol comme solvant : a) CIS1 représente les échantillons de CuInS2 préparés en humidifiant la surface du substrat pendant 3 minutes. b)
