THIBAUT VARIN
MÉTAGÉNOMIQUE DES TAPIS MICROBIENS
POLAIRES
Thèse présentée
à la Faculté des études supérieures et postdoctorales de l’Université Laval dans le cadre du programme de doctorat en physiologie-endocrinologie
pour l’obtention du grade de Philosophiæ doctor (Ph.D.)
DÉPARTEMENT DE MÉDECINE MOLÉCULAIRE FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QUÉBEC
2013
Résumé
Le domaine de l'écologie microbienne est en pleine effervescence grâce à l'avènement de la métagénomique et des techniques de séquençage de nouvelle génération (SNG), qui nous apportent une meilleure compréhension de la structure et du fonctionnement des communautés microbiennes de la biosphère. Cette thèse illustre ainsi une manière de tirer profit de l'utilisation de ces nouvelles technologies, dans le but d'étudier un écosystème qui a été très peu caractérisé jusqu'à maintenant, en l'occurrence les tapis microbiens polaires.
Les analyses métagénomiques de différents tapis microbiens polaires ont permis dans un premier temps, de dresser une description générale de la taxonomie et du potentiel fonctionnel des communautés microbiennes en question, pour ensuite nous permettre d'examiner de façon plus exhaustive deux de leurs particularités métaboliques.
L'existence éventuelle d'un système de recyclage des nutriments au sein même des tapis microbiens étudiés a été soulevée étant donné le caractère oligotrophique de leur milieu environnant. L'analyse des profils métagénomiques des tapis microbiens de l'Arctique a permis de mettre en évidence plusieurs groupes de gènes impliqués dans des mécanismes de décomposition et de récupération qui donneraient la possibilité à ces communautés de retenir et de recycler leurs nutriments au sein de leur microenvironnement benthique. Un autre aspect des tapis microbiens polaires sur lequel je me suis penché lors de ce doctorat, concerne la propension des membres peuplant ce type d'écosystème à s'acclimater à un large panel de stress découlant de la nature extrême de leur habitat. La présence de divers procédés métaboliques d'adaptation au froid et à d'autres stress a été observée à partir de l'analyse du métagénome des ces communautés arctiques et antarctiques, en concordance avec les différents niveaux de représentation des principaux groupes bactériens.
Cette thèse démontre à quel point le recours aux disciplines « méta-omiques », peut nous amener vers une meilleure compréhension de l'écologie microbienne, et comment l'émergence de ces technologies a permis d'aborder différemment des thèmes aussi fondamentaux que celui de la biogéographie des microorganismes.
Abstract
Over the last few years, metagenomics and next generation sequencing (NGS) have been revolutionizing the field of microbial ecology leading to a greater understanding of the structure and functions of the microbial communities in the biosphere. The work presented here applies these new technologies to study polar microbial mats, which are poorly-characterized ecosystems.
Metagenomic analyses of distinct polar microbial mats provided an opportunity to, firstly obtain a general description of microbial community composition and metabolic activity, and subsequently, to more thoroughly study two specific metabolic processes.
We hypothesized that microbial mats are nutrient-replete despite the oligotrophic conditions of the surrounding waters due to strong nutrient recycling within the polar microbial mats. Analyses of metagenomic profiles derived from arctic microbial mats revealed that several groups of genes involved in scavenging mechanisms provide these communities with the capacity to retain and recycle nutrients within the shallow benthic microenvironment.
Another aspect of polar microbial mats which was examined during this PhD, addresses the ability of organisms in the mat to thrive despite varied environmental stresses. The presence of different metabolic processes involved in cold adaptation and other stresses was detected from metagenomic analyses of Arctic and Antarctic communities that were consistently proportional to their representation within major bacterial groups.
This thesis demonstrates how metagenomics and associated « meta-omics » approaches can be informative to improve global comprehension of microbial ecology, and how the emergence of these disciplines enables us to tackle fundamental questions such as biogeography of microorganisms with a new vision.
Avant-propos
En ce qui concerne les contributions apportées lors de ce doctorat, je précise que je suis le premier auteur des deux publications qui sont présentées au chapitre 4 de cette thèse. Plus précisément, je suis responsable de la production intégrale des données et de l'exécution de l'ensemble des analyses bio-informatiques et statistiques nécessaires à la création de la matière sur laquelle repose ces articles. Il faut mentionner cependant, que le travail de laboratoire, qui consistait à réaliser des extractions d'ADN à partir d'échantillons de tapis microbiens polaires, a été exécuté par Anne D. Jungblut. J'ai rédigé la première version de mes deux articles (inclus dans cette thèse), puis retravaillé ensuite leur rédaction sous la supervision de mes coauteurs, à savoir : Connie Lovejoy, Anne D. Jungblut, Warwick F. Vincent, et Jacques Corbeil.
Dans un autre registre, je tiens à remercier Jacques Corbeil pour m'avoir tenté et permis de me lancer dans cette aventure qu'est le doctorat. Je salue également l'ouverture d'esprit, le caractère visionnaire, et la pluridisciplinarité de mon directeur de recherche, qui ont définitivement influencé le déroulement de mes études de troisième cycle d'une manière très positive. Un grand merci, également, à ma codirectrice Connie Lovejoy pour sa gentillesse et sa constante bonne humeur, mais surtout pour ses précieux conseils et son aide dans divers aspects techniques rencontrés durant ce doctorat, que ce soit dans les travaux de laboratoire ou dans la phase de rédaction des mes articles. Ainsi, grâce aux efforts conjugués de mon directeur et ma codirectrice, ce doctorat m'a permis de découvrir le monde de la microbiologie polaire, un domaine passionnant avec lequel je n'étais point familier, et qui m'a sans aucun doute permis d'élargir ma vision du monde microbien. Je tiens à remercier tout particulièrement mes coauteurs Anne D. Jungblut et Warwick F. Vincent, qui m'ont apporté, en plus de leur grande amabilité, une aide indispensable dans l'orientation de mes recherches et lors de la rédaction des articles présentés dans cette thèse. Merci à René Paradis, le pilier du service de bioinformatique au CHUL qui m'a sorti d'innombrable fois du cauchemar que peut représenter un « bug » informatique. Mais au delà du professionnel, je remercie par dessus tout le grand ami, qui est le premier québécois
que j'ai connu en arrivant à Québec, et qui n'a pas hésité à me traiter comme un ami d'enfance. Merci Ron pour ta bonté et ta gaieté si contagieuse ! Merci à Fréderic Raymond, mon voisin de bureau durant toutes ces années passées au CHUL, qui demeure ma personne ressource de référence en génomique, et avec qui je me souviendrai avoir passé de bons moments sur fond de musiques tonitruantes pour reprendre son expression, à l'Impérial, au Velvet, ou encore sur les plaines ! Merci à Sébastien Boisvert pour ses précieux conseils en bio-informatique, ainsi qu'à Robin Allary pour « la » partie de tennis sur l'heure du midi entre deux PCR ! Merci à Pascal Belleau pour son amabilité et ses stimulants sujets de conversations. Je remercie aussi le reste de « l'ancienne » bande du Complan, malheureusement dissoute aujourd'hui; par ordre alphabétique, je pense notamment à Astrid Deschênes, Jean-Philippe Dionne, Arnaud Droit (qui ironie du sort, est un des évaluateurs de cette thèse aujourd'hui), Joël Fillon, Patrick Lacasse, David Paladini, Philippe Rigault, Sayo Yuen.
Merci aux autres membres de l'équipe de Jacques Corbeil, à savoir Lynda Robitaille, Jessyka Fortin, et Nancy Boucher, pour leur aide dans le laboratoire. Je remercie tout particulièrement Mélissa Sirois qui était toujours aussi jovial et serviable même quand je la dérangeais au laboratoire; je suis également reconnaissant envers son conjoint, Yan Sergerie, pour son aide dans la délicate situation de l'après-doctorat.
Du côté du pavillon Vachon ou Marchand, je remercie Estelle Pedneault et Marianne Potvin pour leur disponibilité et leur aide dans les différentes tâches de laboratoire que j'ai eu à exécuter au début de mon doctorat. Merci également à Pierre Galand pour ses conseils avisés dans le domaine de la microbiologie polaire.
Bien évidemment, je remercie de tout cœur ma famille et mes proches, en particulier mes parents et ma sœur qui m'ont toujours aimé, soutenu quoi qu'il arrive, et sans qui je n'aurais pu accomplir toutes les choses que j'ai voulu faire jusqu'à maintenant. Je dédie tout naturellement cette thèse à ma mère qui m'a tant encouragé à faire un doctorat... J'adresse également ma plus sincère reconnaissance à ma « blonde », Kim, qui m'a apporté beaucoup de soutien d'une manière générale tout au long de ce périple, et qui s'est toujours montrée très attentionnée envers moi. Kim a aussi contribué concrètement à cette thèse en m'aidant dans les fastidieuses tâches de relecture et de formatage des références.
Et enfin, après les remerciements adressés aux personnes physiques, je rends hommage à quelque chose de plus abstrait et d'immatériel, en l'occurrence l'escalade ! En effet, ce magnifique sport qui m'a accompagné tout au long de mon doctorat aura été une bouée de sauvetage dans les moments plus difficiles, tant il contribue à l'intégrité de ma santé physique et mentale. De plus, c'est une passion dans laquelle je retrouve beaucoup des valeurs qui me sont chères. J'en profite par la même occasion, pour rendre un dernier hommage à une personne qui m'a beaucoup inspiré en tant que grimpeur mais aussi en tant que personne : Patrick Edlinger, une figure emblématique de l'escalade qui nous a malheureusement quitté alors que je finissais la rédaction de cet avant-propos.
Table des matières
Résumé ... i
Avant-propos ... iii
Table des matières ... ix
Liste des tableaux ... xiii
Liste des figures ... xv
1. Introduction ... 1 1.1. Objectifs ... 3 2. La métagénomique ... 5 2.1. Introduction ... 5 2.1.1. Définition ... 5 2.1.2. Contexte et origine ... 5 2.1.3. Méthodes et applications ... 8 2.1.4. Défis ... 13 2.1.4.1. L'environnement analysé ... 14
2.1.4.2. L'étape d'isolation et d'extraction des acides nucléiques ... 15
2.1.4.3. L'étape de clonage ... 16
2.1.4.4. Le séquençage ... 16
2.2. Séquençage à haut débit ... 17
2.2.1. Historique... 17
2.2.2. Le séquençage en aveugle... 18
2.2.3. Les méthodes à haut débit ... 19
2.2.3.1. Le pyroséquençage 454 ... 20
2.2.3.2. Le séquençage Illumina ... 23
2.2.3.3. Le séquençage ABI SOLiD ... 25
2.2.3.4. Le séquençage par semi-conducteur ... 27
2.2.3.5. Le séquençage par Polonator ... 28
2.2.3.6. Le séquençage HeliScope ... 28
2.2.3.7. Le séquençage SMRT (Single Molecule Real Time) ... 29
2.2.3.8. Le séquençage par Nanopore ... 30
2.3. Métagénomique et bio-informatique ... 32
2.3.1. L'assemblage de génomes ... 32
2.3.1.1. Les assembleurs « voraces » ... 34
2.3.1.2. Les assembleurs OLC ... 35
2.3.1.3. Les assembleurs DBG ... 37
2.3.1.4. L'assemblage de données métagénomiques ... 39
2.3.2. Analyse de biodiversité ... 41 2.3.3. L'annotation de métagénomes ... 43 2.3.3.1. Les défis ... 43 2.3.3.2. L'annotation taxonomique... 44 2.3.3.3. L'annotation fonctionnelle ... 49 2.3.3.4. La métagénomique comparative ... 52 2.3.4. La plateforme MG-RAST ... 54 2.3.4.1. L’historique ... 54
2.3.4.3. La page de vue d'ensemble ... 58
2.3.4.4. La page d'analyse de métagénomes ... 61
2.3.5. L'application STAMP ... 68
2.4. Métagénomique et approches complémentaires... 70
2.4.1. Les tendances futures en termes de métagénomique ... 70
2.4.1.1. La métatranscriptomique ... 70
2.4.1.2. La métaprotéomique ... 73
2.4.1.3. La méta-métabolomique ... 74
2.4.2. Autres approches complémentaires ... 75
3. Les tapis microbiens polaires ... 77
3.1. Définition ... 78
3.2. Les plateformes de glace ... 78
3.2.1. Les plateformes de glace de l'Arctique canadien ... 82
3.2.2. Les tapis microbiens de l'Arctique canadien ... 84
3.2.3. La plateforme de glace de McMurdo (Antarctique)... 85
3.2.4. Les tapis microbiens de la plateforme de McMurdo ... 86
3.3. Composition taxonomique ... 87
3.4. Un écosystème menacé ... 88
4. Analyses métagénomiques de tapis microbiens polaires ... 91
4.1. Le contexte ... 91
4.2. La méthodologie ... 93
4.3. Le recyclage des nutriments au sein des tapis microbiens polaires ... 95
4.3.1. Le résumé en français ... 95
4.3.2. Abstract ... 97
4.4. Article #1 ... 98
4.4.1. Introduction ... 99
4.4.2. Methods ... 100
4.4.2.1. Study sites and sample collection... 100
4.4.2.2. DNA extraction and sequencing ... 101
4.4.2.3. Bioinformatics and statistical analyses ... 102
4.4.3. Results ... 103
4.4.3.1. Classification of microbial communities based on protein-coding genes . 104 4.4.3.2. Taxonomic classification of the microbial communities based on ribosomal genes ... 105
4.4.3.3. Functional gene analysis ... 106
4.4.4. Discussion ... 108
4.4.5. Acknowledgments ... 113
4.5. Tables ... 114
4.6. Figures ... 118
4.7. Stress et tapis microbiens polaires ... 123
4.7.1. Le résumé en français ... 123
4.7.2. Abstract ... 125
4.8. Article #2 ... 126
4.8.1. Introduction ... 127
4.8.2. Materials and Methods ... 128
4.8.2.1. Study site and sample collection ... 128
4.8.2.3. Bioinformatics and statistical analyses ... 129
4.8.2.4. Metagenome sequence accession ... 131
4.8.3. Results ... 131
4.8.3.1. Mat metagenomes ... 131
4.8.3.2. Taxonomic and functional comparisons of polar microbial mats ... 132
4.8.3.3. Taxonomy of genes involved in cold and other stresses ... 133
4.8.3.4. Taxonomy of functional differences ... 134
4.8.4. Discussion ... 135
4.8.5. Acknowledgments ... 140
4.9. Tables ... 141
4.10. Figures ... 149
5. Discussion ... 153
5.1. Notre approche métagénomique et ses alternatives ... 153
5.1.1. L'approche utilisée dans nos articles ... 153
5.1.2. Une alternative à notre approche ... 155
5.1.3. Des exemples d'analyses complémentaires ... 165
5.2. Biogéographie microbienne des habitats de froid pérenne ... 167
5.2.1. Distribution microbienne au sein des tapis microbiens polaires... 167
5.2.2. Hypothèses d'écologie microbienne et habitats de froid pérenne ... 169
5.2.2.1. L'hypothèse de l'ubiquité ... 170
5.2.2.2. L'endémisme microbien ... 175
5.2.3. Combinaison de cosmopolitisme et d'endémisme ? ... 177
6. Conclusion ... 183
Liste des tableaux
Tableau 2.1. Liste (non exhaustive) d'études pertinentes retraçant l'histoire de la métagénomique (tableau adapté de Ederer, 2011). Ces différents projets sont classés selon le type de communautés microbiennes analysées. Abréviations : Gbases (gigabases), Mbases (mégabases), 454 (pyroséquençage 454), ABI (séquençage ABI SOLiD), Illumina GA (séquençage Illumina Genome Analyzer), Illumina HS (séquençage Illumina HiSeq
2000). ... 12
Table 4.5.1. Temperature, pH, conductivity of the meltwater ponds for the three samples that were pooled for each metagenome. MIS,
Markham Ice Shelf; WHI, Ward Hunt Ice Shelf. ... 114
Table 4.5.2. Percentage of total significant hits found from combined MIS and WHI sequences when compared to databases designated in the top row. Taxonomic analysis based on rRNA genes was performed with BLASTN against RDP II and Euro rRNA SSU databases. SEED and Swiss-Prot were used with BLASTX indicating taxonomic placements based on protein-coding genes. E-value cut-off: 1 × 10−5 for all. Total of significant hits
from both MIS and WHI sequences; na, not applicable. ... 115
Table 4.5.3. Sequence matches to Eukaryota obtained using the MG-RAST server against Euro rRNA SSU database (E-value cutoff: 0.1). Positive hits to the SCUM database for prokaryote viral sequences and GenBank for eukaryote viral sequences (E-value
cutoff: 1 × 10−4). ... 116
Table 4.9.1. Environmental and metagenomic comparisons for the three
sampling sites ... 141
Supplementary Table 4.9.4. Significant matches found to microbial polar
genomes and the model cyanobacterial reference genome Synechococcus WH5701. Comparisons among the metagenomes from the Markham Ice Shelf (MIS), Ward Hunt Ice Shelf (WHI) and McMurdo Ice shelf (MCM) using BLASTX compared to the SEED database to indicate bacterial or archaeal taxonomic placements based on protein-coding genes (E value ≤10-5, alignment length ≥50 bp; percentage of identity >65%). Values were normalized to the metagenome (WHI) with the highest amount of BLASTX hits. NCBI reference sequences of genomes are indicated in parentheses.
Liste des figures
Figure 2.1. Schématisation de plusieurs méthodologies courantes dans une analyse métagénomique (figure adaptée d'Ederer, 2011). Le chemin constitué de cadres et de flèches rouges symbolise la méthode métagénomique utilisée dans les différentes analyses
présentées dans cette thèse. ... 9
Figure 2.2. Principe du pyroséquençage 454 (figure adaptée de 454 Life Sciences, © 1996-2012 Roche Diagnostics Corporation,
www.454.com). ... 22
Figure 2.3. Principe du séquençage Illumina (figure tirée de Strausberg et
al., 2008). ... 24
Figure 2.4. Principe du séquençage ABI SOLiD® (figure tirée de
Strausberg et al., 2008). ... 26
Figure 2.5. Sources de la distribution des résultats significatifs obtenus avec MG-RAST pour l’échantillon métagénomique de tapis microbien prélevé sur la plateforme de glace de Ward Hunt. Ce graphique illustre le nombre de fragments appartenant à ce métagénome qui ont obtenu des résultats significatifs par similarité de séquence avec les différents types de bases de données cités dans la présente figure (protéiques, protéiques hiérarchisées, ou ribosomiques). Une échelle de couleurs permet de symboliser l'étendue d'« E-value » utilisée pour
chaque base de données. ... 59
Figure 2.6. Menu de la page d'analyse de MG-RAST. L'encadré rouge intitulé « Data Type » permet à l'utilisateur de choisir son type d'analyse taxonomique (méthode du meilleur résultat significatif obtenu, ou du plus petit ancêtre commun) ou fonctionnelle (selon une classification hiérarchique, ou selon toutes les bases de données fonctionnelles hiérarchisées ou non), ou encore de lancer une analyse de recrutement. L'encadré bleu nommé « Data Selection » permet de choisir le (ou les) métagénome(s) à analyser (ou comparer), la base de données à utiliser, ainsi que la valeur des paramètres à considérer pour l'analyse (« E-value » maximum, pourcentage d'identité minimum, et longueur d'alignement minimum). L'encadré mauve appelé « Data Visualization » propose à l'utilisateur de choisir le mode de visualisation des résultats en fonction de l'analyse à effectuer (graphique en barres, arbre phylogénétique, tableau, carte phylogénétique (« heatmap »), graphique en composantes principales, ou courbe de raréfaction). Dans cet exemple, l'utilisateur désire réaliser une analyse taxonomique du métagénome 4445129.3 (échantillon de tapis microbien
prélevé sur la plateforme de glace de Ward Hunt) basée sur la classification à partir du meilleur résultat significatif obtenu. L'utilisateur a sélectionné la base de données M5NR et a laissé les autres paramètres par défaut, de plus, en sélectionnant l'onglet « table », il a choisi d'afficher les résultats de l'analyse
dans un tableau (du type de celui de la Figure 2.7.). ... 62
Figure 2.7. Tableau interactif généré par MG-RAST qui représente les résultats significatifs obtenus pour une analyse taxonomique du métagénome 4445129.3 (échantillon de tapis microbien prélevé sur la plateforme de glace de Ward Hunt). Dans cet exemple, il s'agit d'une analyse de classification taxonomique basée sur la similarité en fonction du meilleur résultat significatif obtenu (« best hit classification ») contre la base de données protéiques M5NR, en tenant compte des paramètres suivants : « E-value » maximum de 1.10-5, pourcentage d'identité minimum de 65%, et longueur d'alignement minimum de 50 pb. Chaque ligne du tableau indique dans une colonne distincte de gauche à droite respectivement : l'identifiant du métagénome en question, la base de données utilisée, le rang taxonomique avec une colonne par niveau taxonomique (du domaine à la souche selon le choix de l'utilisateur), l'abondance (nombre de séquences du métagénome qui ont été assignés à l'annotation en question), la valeur moyenne des « E-values », des pourcentages d'identité, et des longueurs d'alignement des appariements significatifs trouvés, et enfin le nombre de « hits » (nombre de séquences uniques appartenant à la base de données en question qui ont eu un appariement significatif). À noter que dans cet exemple de tableau, seules les 25 premières lignes (sur un total de 94) apparaissent; de plus, ici l'utilisateur aura choisi préalablement d'afficher uniquement les domaines, phyla, et classes de chaque organisme. Les nombres indiqués en bleu dans la colonne de l'abondance sont cliquables et permettent d'accéder à une nouvelle page où sont affichés tous les détails de l'alignement correspondant aux résultats présentés dans la ligne en question. Deux onglets situés en haut à droite du tableau, appelé « krona graph » et « QIIME report » permettent respectivement d'afficher les données du tableau sous la forme d'un graphique de type « krona », ou de télécharger localement ces mêmes
données dans un format compatible avec QIIME. ... 64
Figure 3.1. Plateforme de glace de Ward Hunt, située le long de la côte nord de l’île d’Ellesmere, au Nunavut, dans le haut Arctique
canadien. (Photographe: Warwick F. Vincent, 2006) ... 79
Figure 3.2. Tapis microbien recouvert partiellement d’eau de fonte sur la plateforme de glace de Markham, située sur la côte nord de l’ile
d’Ellesmere, au Nunavut, dans le haut Arctique canadien.
(Photographe: Anne D. Jungblut, 2006) ... 80
Figure 3.3. Gros plan d'un tapis microbien affichant une pigmentation orangée caractéristique, et gisant au fond d'une marre d'eau de fonte présente sur la plateforme de glace de Ward Hunt.
(Photographe: Warwick F. Vincent, 2006) ... 81
Figure 3.4. Tapis microbien de la plateforme de Markham exposés à l'air et présentant une forte pigmentation orangée en surface.
(Photographe: Warwick F. Vincent, 2006) ... 81
Figure 3.5. Gros plan d’un échantillon de tapis microbien prélevé sur la plateforme de glace de Markham. (Photographe: Anne D.
Jungblut, 2006) ... 85
Figure 4.6.1. Location of sampling sites along the northern coast of Ellesmere Island in High Arctic, Canada. Dark gray indicates the extent of the two ice shelves at the time of sampling (2006);
the white circles indicate sites where the mats were collected... 118
Figure 4.6.2. Inferred taxonomic distributions from the MIS (black bars) and WHI (grey bars) microbial mat metagenomes. Sequence classification based on BLASTX similarities to SEED from protein-coding genes. Insert: contribution of the major classes of Proteobacteria (A = Alphaproteobacteria; B = Betaproteobacteria; G = Gammaproteobacteria; O = other
Proteobacteria). Note the log scale of the y-axis in both graphs. ... 119
Figure 4.6.3. Percentage of sequences assignable to functional categories for MIS (black bars) and WHI (gray bars) metagenomes. Sequence groupings in subsystem (ss) categories were performed by the MG-RAST server using the SEED database. Note the log scale
of the y-axis. ... 120
Figure 4.6.4. Principal component analysis of gene functions from different bacterial groups. (A) Genes involved in light capture. (B) Nitrogen uptake and transformation genes. (C) Phosphorus uptake and transport genes. Horizontal and vertical axes represent component 1 and component 2, respectively. Gray circles contain all other identified taxa. Abbreviations: PSI, photosystem I; PSII, photosystem II; PSII-like, bacterial photosystem II–type photosynthetic reaction center; Nfix, nitrogen fixation; AmmAss, ammonium assimilation system; Denitrif, denitrification; AllantoinD, allantoin degradation; UreaD, urea degradation; CyanateH, cyanate hydrolysis; Na and NiAmo, nitrate and nitrite reduction to ammonia; ChitinN, chitin and N-acetylglucosamine utilization; Cyanoph, cyanophycin metabolism; ABCbcaa, ABC transporter for branched-chain amino acids; ABCdip, ABC transporter for dipeptides; ABCgluta, ABC transporter for glutamate and
aspartate; ABCglut, ABC transporter for glutamine; ABColigo, ABC transporter for oligopeptides; Alkalin_Pho, alkaline phosphatase; PhoTransPhoReg, high-affinity phosphate transporter and control of PHO regulon; ExoPho, exopolyphosphatase; Polypho, polyphosphate kinase; AlkylphosphonateU, alkylphosphonate utilization; pstA, phosphate transport system permease protein pstA; pstB, phosphate transport system permease protein pstB; pstC, phosphate transport system permease protein pstC; pstS, phosphate ABC transport, periplasmic phosphate-binding protein pstS; phoU, phosphate transport regulator phoU; PhoR, phosphate regulon sensor protein phoR; phoB, phosphate
regulon transcriptional regulatory protein phoB. ... 121
Figure 4.10.1. Statistical analyses of taxonomic profiles for the Arctic (combined MIS and WHI samples) and Antarctic (MCM sample) metagenomes. Orders or classes overrepresented in the Antarctic have a negative difference between proportions (green dots); those overrepresented in the Arctic community have a positive value difference between proportions (blue dots). Features (orders or classes) with a q value of ˃0.05 were
considered biologically significant. ... 149
Figure 4.10.3. Statistical analyses of metabolic profiles for the Arctic microbial mats (combined MIS-WHI samples) and the Antarctic metagenome (MCM sample). Total numbers of sequences in the different categories are shown in the left bar graph; the left side (blue) represents the Arctic mats, while the right side (green) represents the Antarctic mat. Subsystems in the Antarctic microbial mat community have negative differences between proportions (green dots). Subsystems overrepresented in the Arctic microbial mat samples have positive differences between proportions (blue dots). Features (orders or classes) with a q value of >0.05 were considered
significant. ... 152
Figure 5.1. Nombres de séquences (n) appartenant aux métagénomes arctiques (MIS (en rouge) et WHI (en gris)) et antarctiques (MCM (en bleu)) qui ont pu être assignées à différents gènes impliqués dans les réactions biochimiques du cycle de l'azote. L'épaisseur des flèches est proportionnelle au niveau d'abondance relative de séquences assignées aux gènes impliqués dans la réaction en question. Ces annotations fonctionnelles ont été réalisées en utilisant BLASTX contre la base de données SEED; seuls les appariements significatifs ayant plus de 50 nucléotides de longueur, au moins 65% de similarité, et un « E value » ≤10-5, ont été inclus. Les valeurs (n)
ont été normalisées par rapport au métagénome (WHI) ayant
obtenu le plus de « hits » avec BLASTX. ... 159
Figure 5.2. Graphique d'analyse en coordonnées principales représentant le degré de similitude taxonomique entre différents métagénomes de tapis microbiens provenant de divers environnements (polaires, chauds, et tempérés). Abréviations : MIS : Markham Ice Shelf microbial mat, WHI : Ward Hunt Ice Shelf microbial mat, MCM : McMurdo Ice Shelf microbial mat; AL : Antarctic freshwater lake (Ace lake, Antarctica); MHS : Mushroom hot spring microbial mat (Yellowstone National Park), OHS : Octopus hot spring microbial mat (Yellowstone National Park); GMM : Green freshwater microbial mat (Cuatro Ciénegas Basin II, Coahuila, Mexico), RMM : Red freshwater microbial mat (Cuatro Ciénegas Basin II, Coahuila, Mexico). Pour chaque métagénome, les séquences ont été annotées taxonomiquement à l'aide de BLAT contre la base de données M5NR, en considérant comme significatif les appariements ayant plus de 50 nucléotides de longueur, au moins 65% de similarité, et un «
E value » ≤10-5. ... 163
Figure 5.3. Graphique d'analyse en coordonnées principales représentant le degré de similitude fonctionnelle entre différents métagénomes de tapis microbiens provenant de divers environnements (polaires, chauds, et tempérés). Abréviations : MIS : Markham Ice Shelf microbial mat, WHI : Ward Hunt Ice Shelf microbial mat, MCM : McMurdo Ice Shelf microbial mat; AL : Antarctic freshwater lake (Ace lake, Antarctica); MHS : Mushroom hot spring microbial mat (Yellowstone National Park), OHS : Octopus hot spring microbial mat (Yellowstone National Park); GMM : Green freshwater microbial mat (Cuatro Ciénegas Basin II, Coahuila, Mexico), RMM : Red freshwater microbial mat (Cuatro Ciénegas Basin II, Coahuila, Mexico). Pour chaque métagénome, les séquences ont été annotées taxonomiquement à l'aide de BLAT contre la base de données SEED, en considérant comme significatif les appariements ayant plus de 50 nucléotides de longueur, au moins 65% de similarité, et un «
1. Introduction
Malgré le fait que, techniquement parlant, la génomique a permis de réaliser des progrès fulgurants dans le domaine de l’écologie microbienne, il faut attendre le début des années 2000, avec l’arrivée de la métagénomique, pour qu’une véritable révolution s’opère dans le monde de la microbiologie environnementale. Durant les dernières années, la métagénomique s’est taillée une place de choix dans le palmarès des disciplines scientifiques les plus actives et les plus utilisées par la communauté scientifique. Cette approche expérimentale novatrice et particulièrement puissante, permet désormais l’analyse de communautés microbiennes qui semblaient largement hors de portée il y a moins de dix ans. Le fait de pouvoir accéder à l’information génétique d’un assemblage microbien directement depuis son environnement naturel, tout en s’affranchissant de certaines contraintes liées à la microbiologie traditionnelle, a ouvert la voie à une nouvelle dimension dans l'exploration des écosystèmes microbiens.
Depuis les trente dernières années, un autre domaine est en plein essor, c'est celui de la microbiologie polaire. L'amélioration de l'accessibilité à ces régions isolées a provoqué un véritable engouement auprès des microbiologistes, qui se sont empressés d'approfondir notre savoir sur l'écologie des communautés microbiennes vivant dans ces environnements de froid intense. D'autant plus qu'une meilleure compréhension des écosystèmes polaires serait susceptible d'apporter de plus amples connaissances sur des sujets aussi fondamentaux que l'apparition de la vie sur Terre, les cycles biogéochimiques, ou encore les changements environnementaux à l'échelle planétaire, qu'ils soient passés ou actuels.
Durant ce doctorat, j'ai choisi de coupler la métagénomique et la microbiologie polaire, afin d'apporter ma modeste contribution dans l'étude d'un écosystème très peu exploré jusqu'à présent, à savoir les tapis microbiens des régions polaires. Mes travaux avaient donc pour but d'évaluer certaines caractéristiques physiologiques inhérentes à la survie de ces communautés microbiennes dans un habitat particulièrement hostile.
Faisant suite à la présente introduction, le deuxième chapitre de cette thèse dresse un portrait technique de cette fantastique discipline qu'est la métagénomique, puis décrit ensuite les technologies complémentaires sans lesquelles la métagénomique ne pourrait être utilisée efficacement, il s'agit en l'occurrence des techniques de séquençage à haut débit et des différents outils bio-informatiques de traitements des données. Le chapitre 2 est le plus conséquent de ma thèse car il est représentatif de l'importance des choix en matière de séquençages à haut débit, d'approches métagénomiques, et de méthodes d'annotations. De plus, je voulais que ce chapitre soit à l'image de l'ampleur du travail que j'ai fourni lors de ce doctorat dans la considération de ces différents aspects, afin de mener à bien nos objectifs.
Le chapitre 3 est quant à lui dédié à la description générale des tapis microbiens polaires en tant que tels et de leur habitat structurel qui est constitué par les plateformes de glaces. Ce chapitre est conclu par une note plutôt alarmiste qui met en évidence l'urgence d'étudier un tel écosystème avant qu'il ne disparaisse en raison du contexte de réchauffement climatique actuel.
On entre dans le vif du sujet à proprement parler avec le chapitre 4, qui présente les deux publications réalisées durant ce doctorat, qui traitent d'une part du recyclage des nutriments au sein des tapis microbiens polaires, et d'autre part des mécanismes d'adaptation à différents stress utilisés par ces communautés microbiennes.
La discussion énoncée dans le chapitre 5 aborde dans un premier temps les tendances futures en termes de métagénomique, avec notamment la description des différentes disciplines « méta-omiques » ainsi que leurs applications possibles. J'évoque ensuite les forces et les faiblesses de l'approche métagénomique utilisée dans nos articles, pour ensuite proposer une approche méthodologique alternative, ainsi que des exemples d'analyses complémentaires qui permettraient de compléter les travaux réalisés lors de ce doctorat. Dans une deuxième partie, j'aborde un thème aussi passionnant que complexe, celui de la biogéographie microbienne au sein de la cryosphère. Je compare ainsi certains arguments en faveur de l'une ou l'autre des deux plus grandes théories liées à la répartition planétaire
des microorganismes, c'est-à-dire le cosmopolitisme et l'endémisme, tout en étayant chacune de ces notions à l'aide de données issues de nos propres analyses ou provenant d'autres publications sur le sujet.
1.1. Objectifs
Face au caractère plutôt modeste des connaissances actuelles sur les tapis microbiens polaires, le but de mes travaux de doctorat était d'utiliser la puissance de la métagénomique afin de tenter de mieux comprendre la structure et les processus métaboliques utilisés par ces communautés microbiennes.
Plus précisément, l'objectif général était de réaliser, à partir de métagénomes de tapis microbiens récoltés sur diverses plateformes de glace polaires, des profils fonctionnels et taxonomiques de basse résolution, afin d'obtenir une vue d'ensemble de la composition et du potentiel métabolique des tapis microbien polaires, tout en s'efforçant de comparer cette dernière en fonction de leur origine (Arctique ou Antarctique).
De plus, ce type d'approche devait nous permettre de définir les deux objectifs spécifiques suivants :
Mettre en évidence la présence d'un système de décomposition et de recyclage des nutriments au sein des tapis microbiens de l'Arctique, les rendant ainsi capables de former un microenvironnement riche en nutriments malgré le caractère oligotrophique de leur habitat;
Évaluer si les similarités taxonomiques éventuelles entre les tapis microbiens arctiques et antarctiques se reflètent sur leurs réponses génétiques face aux stress environnementaux. Ceci permettant de tester par la même occasion, si des communautés microbiennes occupant le même type d'habitat extrême déploient les mêmes types de mécanismes de résistance et d'adaptation aux stress imposés par l'environnement polaire, et ceci indépendamment de la distance géographique qui les sépare.
2. La métagénomique
De par l'introduction, vous aurez compris que la métagénomique revêt une importance capitale dans cette thèse; c'est la raison pour laquelle je lui dédie ce chapitre, qui a pour but de familiariser le lecteur avec les différents aspects, principes et applications de la métagénomique.
2.1. Introduction
2.1.1. Définition
La métagénomique consiste à analyser l'ADN génomique d'une communauté microbienne dans son ensemble. En d'autres mots, c'est une approche basée sur l'isolation directe de l'intégralité des acides nucléiques présents dans un échantillon prélevé dans un environnement donné, et ceci sans aucun isolement ou culture de microorganismes au préalable (Handelsman, 2004; Simon & Daniel, 2011). Le préfixe « méta » qui en grec veut dire littéralement « au-delà », induit une distinction majeure entre les termes « métagénomique » et « génomique », ce dernier représentant l'étude de l'ADN génomique issu d'un seul microorganisme ou d'une cellule unique (Gilbert & Dupont, 2011).
2.1.2. Contexte et origine
Durant la majeure partie de son histoire, la microbiologie est restée centrée sur le fait que la culture de microorganismes était une étape inhérente à toutes études dignes de ce nom. En effet, les microbiologistes ont longtemps cru que les organismes microbiens pouvaient être classifiés, si et seulement si, ils étaient cultivés auparavant (Society of American Bacteriologists, 1923). Cependant, dans les années 1980, les scientifiques ont réalisé que les microbes étaient beaucoup plus ubiquitaires, diversifiés, et nombreux que ce qu'ils pensaient (Ederer, 2011); et nous savons désormais que les techniques classiques de culture utilisées en microbiologie ont permis jusqu'à maintenant de caractériser seulement environ 1% de la diversité microbienne planétaire (D'Onofrio et al., 2010). Des estimations calculées par Whitman et collaborateurs (Whitman et al., 1998) indiquent que la terre est l'habitat d'environ 5×1030 procaryotes (bactéries et archées) avec plus de 107 espèces de
bactéries différentes, ces dernières étant présentes dans tous les environnements planétaires, capables de coloniser les habitats les plus extrêmes allant des sources hydrothermales à 340°C des profondeurs océaniques, aux tapis microbiens polaires que nous avons étudiés (Whitman et al., 1998).
Mais pourquoi l'étude de l'écologie microbienne devrait susciter autant d'intérêt ?
Les microorganismes ne sont pas seulement omniprésents, ils sont essentiels à la vie sur terre car ils représentent le plus grand réservoir de nutriments de la planète en séquestrant 50% du carbone total de tous les organismes vivants et en constituant le plus grand réservoir terrestre de nitrogène et de phosphore cellulaire (Whitman et al., 1998). De plus, les microbes sont les principaux recycleurs de matières en décomposition permettant de rendre à nouveau disponible divers composés sous forme organique (Whitman et al., 1998). Ils jouent donc un rôle critique dans les processus biogéochimiques en maintenant la survie des écosystèmes (Falkowski et al., 2008). Par ailleurs, l'étude de la diversité microbienne des environnements extrêmes, tels que les sources chaudes (Rhee et al., 2005), les bassins hyper-salins (Ferrer et al., 2005), la glace de glacier (Simon et al., 2009), et les sols ou sédiments arctiques et antarctiques (Heath et al., 2009; Jeon et al., 2009), suscite un certain engouement étant donné du fait que ces écosystèmes représentent un réservoir de nouvelles biomolécules ayant potentiellement un intérêt au niveau biotechnologique.
Tout comme les plantes et les autres animaux, l'existence même de l'homme est fortement dépendante et affectée par les microorganismes. Nous avons plus de cellules bactériennes (1014) résidant dans notre corps que de cellules humaines (1013) (Berg, 1996). Le matériel génétique de l'ensemble de ces bactéries représente environ 1000 fois plus de gènes que ceux du génome humain (Ederer, 2011). Par ailleurs, Turnbaugh et collaborateurs (Turnbaugh et al., 2006) ont déjà démontré que des différences dans la composition du microbiome humain peuvent être liées à certaines conditions pathologiques chez l'homme. En résumé, on peut affirmer que l'étude de l'écologie des communautés microbiennes de la biosphère constitue une première étape fondamentale dans la compréhension :
De leurs contributions dans le maintien de la santé planétaire;
De leurs rôles dans le bien-être chez l'humain;
Des conséquences environnementales de l'activité humaine.
Au vu du peu de connaissances de la communauté scientifique en termes de biodiversité microbienne, les microbiologistes avaient besoin de nouvelles approches moins réductrices, et indépendantes de la culture pour la caractérisation des microorganismes, prenant ainsi en compte l'écosystème dans son ensemble. Le développement de nouvelles méthodologies devaient permettre ainsi de caractériser les organismes identifiés dans leur environnement, ceci impliquant de pouvoir déceler leurs possibles interactions avec les autres membres de la communauté et leur potentiel métabolique.
C'est dans ce contexte que Pace et collaborateurs (Pace et al., 1986), qui aspiraient à étudier la diversité et la classification des bactéries par d'autres méthodes que celles basées sur les traits morphologiques et physiologiques des ces organismes, furent les premiers à analyser directement (sans culture au préalable) les gènes codant pour l'ARN ribosomique (ARNr) 16S présents dans un échantillon environnemental afin de déterminer sa diversité microbienne. Depuis l'étude novatrice de Pace et collaborateurs en 1986 (Pace et al., 1986), l'utilisation de l'ARNr 16S (couplée à la technologie de séquençage de Sanger (Sanger et al., 1977) déjà très populaire à l'époque) s'est rapidement imposée au fil des années comme outil de prédilection pour la discrimination des différentes espèces bactériennes. Ce sont donc des recherches de ce type qui ont permis ultimement la naissance de la métagénomique, c'est-à-dire, une discipline nouvelle qui se situe à l'interface de la microbiologie, la génomique et l'écologie (Ederer, 2011). Ce terme ayant été mentionné officiellement pour la première fois par Handelsman et collaborateurs (Handelsman et al., 1998) lors d'une étude visant à découvrir des nouveaux antibiotiques à partir de communautés microbiennes vivant dans le sol.
Cette nouvelle approche qu'est la métagénomique vise ainsi, en analysant le contenu en acides nucléiques d'une communauté microbienne quelconque, à caractériser la diversité microbienne, la complexité génétique, mais aussi ses diverses interactions dont celles de
nature métabolique. C'est sur ce dernier point que la métagénomique se distingue, en permettant d'aller bien plus loin que les analyses basées seulement sur l'amplification PCR des gènes codant pour l'ARNr 16S ou 18S qui permettent de se concentrer uniquement sur des études taxonomiques. Ainsi, avec le soutien essentiel de la bio-informatique, le couplage de la métagénomique avec le séquençage à haut débit, offre dorénavant la possibilité d'accéder à des domaines inexplorés du monde microbien.
La complexité et le nombre sans cesse grandissant de projets métagénomiques ont engendré la création par la communauté scientifique de consortiums visant à aider les chercheurs utilisant des données métagénomiques, en offrant des cyber-infrastructures permettant essentiellement le catalogage des génomes (complets) de référence issus de métagénomes et des outils d'analyse et de consultation de ces données. On peut distinguer à l'heure actuelle quatre principales ressources de ce type : le « Human Microbiome Project (HMP) » (http://www.hmpdacc.org/), le « Global Ocean Sampling (GOS) expedition » (http://www.jcvi.org/cms/research/projects/gos/), le « Terragenome » (http://www.terragenome.org/), et enfin mais non le moindre, le « Earth Microbiome Project (EMP) ».
2.1.3. Méthodes et applications
La métagénomique représente un attrait indéniable pour les scientifiques dans le sens où elle leur permet d'accéder à un certain nombre des membres d'une communauté microbienne donnée (indépendamment du fait de savoir si ces organismes peuvent être cultivés ou pas), tout en ayant la possibilité d'accéder à une vue d'ensemble des voies métaboliques régnant en son sein.
Ceci étant dit, quels sont les types d'approches métagénomiques et leurs principales étapes en termes de protocole ?
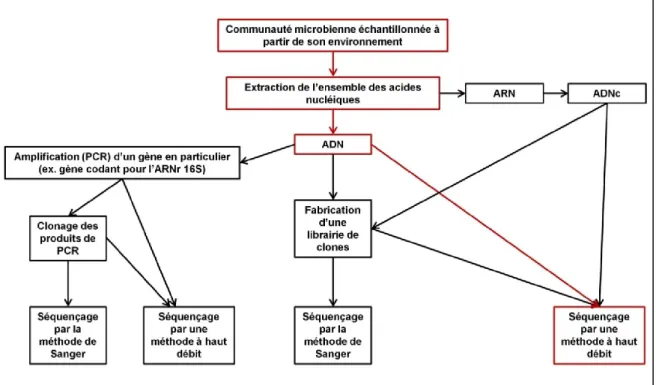
Cela dépend bien évidemment du but de l'étude en question mais on peut résumer ces étapes à la manière de celles illustrées dans la figure 2.1.
Figure 2.1. Schématisation de plusieurs méthodologies courantes dans une analyse
métagénomique (figure adaptée d'Ederer, 2011). Le chemin constitué de cadres et de flèches rouges symbolise la méthode métagénomique utilisée dans les différentes analyses présentées dans cette thèse.
Dans tous les cas, on doit procéder après échantillonnage de l'écosystème en question, à une extraction de l'ensemble de l'ADN ou l'ARN génomique présent dans le ou les échantillon(s). Si l'estimation de la diversité microbienne est le seul but de l'étude en question, une amplification PCR spécifique des gènes codant pour l'ARNr 16S ou 18S sera réalisée afin de séquencer uniquement ces derniers en bout de ligne; on obtiendra ainsi une librairie à grande échelle de gènes d'ARN ribosomique. Une telle librairie peut ensuite être soumise à une biopuce de type PhyloChip afin de pouvoir comparer la diversité de cette dernière avec un ensemble de gènes codant pour des ARNr 16S connus (Brodie et al., 2006; DeSantis et al,. 2007; Yergeau et al., 2009). Par contre, si on s'intéresse cette fois au potentiel métabolique d'un écosystème, on va s'efforcer d'analyser l'ensemble des acides nucléiques de l'échantillon. L'ADN ou l'ADNc (ADN complémentaire obtenu par rétrotranscription de l'ARN isolé) extrait peut être alors traité essentiellement de trois manières différentes :
Il peut être fragmenté aléatoirement, puis séquencé directement en utilisant des technologies de séquençage à haut débit. Il faut noter que ce séquençage direct d'acides nucléiques après extraction est de plus en plus privilégié à l'heure actuelle, étant donné qu'il permet de se soustraire à toute étape de clonage. À noter cependant que la plupart des techniques de séquençage à haut débit nécessitent la création d'une librairie de fragments d'ADN, néanmoins ces derniers ne sont pas clonés, et il ne faut donc pas confondre ce type de librairie avec celui mentionné dans le paragraphe suivant;
Après fragmentation aléatoire, l'ADN peut également être cloné afin de constituer une librairie de clones à petits ou larges inserts qui sera ensuite séquencée par la technique de Sanger (Sanger et al., 1977) ou par une technologie à haut débit; c'est ce qu'on appelle le séquençage en aveugle (voir section 2.2.2.). Cette librairie peut ensuite être parcourue pour rechercher puis séquencer des clones comportant un gène d'intérêt, ou bien séquencée en totalité pour permettre la reconstitution de génomes complets (Venter et al., 2004);
Troisièmement, l'ADN génomique extrait peut aussi être analysé par des biopuces de type GeoChip 3.0 contenant un ensemble de sondes spécifiques à des marqueurs phylogénétiques (tel que gyrB) et à des gènes d'intérêts connus (appartenant à des cycles biogéochimiques par exemple) (He et al., 2007; 2010).
Dans tous les cas, le volume conséquent de fragments séquencés (appelés « reads » en anglais) lors d'une étude de métagénomique, fait en sorte que l'utilisation de la bio-informatique est indispensable afin d'analyser ces derniers.
D'un point de vue général, toute étude faisant appel à la métagénomique doit s'orienter vers l'une des approches suivantes (DeLong, 2007) :
La première est centrée sur le « génome » afin de déterminer les membres composant une communauté microbienne, avec le but ultime de tenter d'assembler leur génome complet;
La seconde aspire quant à elle, à réaliser une analyse fonctionnelle de la communauté échantillonnée afin de déceler son potentiel métabolique. Elle est donc basée cette fois-ci non pas sur le « génome » mais sur le « gène ». C'est d'ailleurs cette approche qui a été privilégiée dans le cadre de cette thèse pour l'étude des tapis microbiens polaires.
La combinaison des deux approches citées précédemment est également envisageable.
Les études métagénomiques effectuées jusqu'à présent ont porté sur trois principaux types de communautés microbiennes :
Les communautés que l'on retrouve à l'état naturel (environnements tempérés ou extrêmes);
Les communautés vivant dans un environnement modifié par l'activité humaine; Les communautés qui dépendent d'un hôte pour survivre.
Le tableau 2.1. recense un certain nombre d'exemples d'études métagénomiques classées selon les trois catégories de communautés microbiennes citées précédemment. Cette liste n'est en aucun cas exhaustive, elle a seulement pour but d'illustrer la grande diversité des projets de métagénomique déjà réalisés à ce jour.
Tableau 2.1. Liste (non exhaustive) d'études pertinentes retraçant l'histoire de la
métagénomique (tableau adapté de Ederer, 2011). Ces différents projets sont classés selon le type de communautés microbiennes analysées. Abréviations : Gbases (gigabases), Mbases (mégabases), 454 (pyroséquençage 454), ABI (séquençage ABI SOLiD), Illumina GA (séquençage Illumina Genome Analyzer), Illumina HS (séquençage Illumina HiSeq 2000).
Auteurs Objectifs de l'étude métagénomique Taille du projet Type de séquençage
Communautés à l'état naturel Hugenholtz et al., 1998 Taxonomie bactérienne d'une source chaude du Yellowstone Librairie de gènes d'ARNr (˃300 clones) ABI Rondon et al., 2000 Identification de nouveaux gènes à partir de microbiomes du sol
1 Gbases clonées dans
des BACs ABI
Breitbart et al., 2002 Comparaison de deux communautés océaniques de virus
Librairie de gènes d'ARNr 16S clonés
(˃1000 clones) ABI Venture et al., 2004 Microbes de la mer des Sargasses 1,045 Gbases clonées ABI Edwards et al., 2006 Population microbienne de la mine « Soudan »
aux États-Unis 70 Mbases 454 Rusch et al., 2007 Échantillonnage global de l'océan (GOS) 6,4 Gbases clonées ABI Dinsdale et al., 2008
Métagénomique des profils métaboliques de 9
biomes 15 Mbases 454
Breitbart et al., 2009 Communautés microbiennes associées
aux microbialites 47,1 Mbases 454 Varin et al., 2010;
2012
Les tapis microbiens
polaires 146 Mbases 454 Communautés affectées par l'activité humaine Martin et al., 2006 Taxonomie des communautés de boue activée dans le traitement des eaux usagées
176 Mbases ABI Thurber et al., 2009 Facteurs de stress sur le corail holobionte 22 Mbases 454 Tamaki et al., 2011 Virus à ADN contenus dans les eaux usées 70-119 Mbases 454
Yu & Zhang, 2012
Métagénomique et métatranscriptomique de boue activée dans le traitement des eaux usagées
Tableau 2.1. (suite)
Auteurs Objectifs de l'étude métagénomique Taille du projet Type de séquençage
Communautés hôte-dépendantes
Breitbart et al., 2003 Communauté virale des excréments humains 17 Mbases d'ADN contenues dans une
librairie de 532 clones ABI Gill et al., 2006 Métagénomique de la partie distale de l'intestin 78 Mbases avec 2062 clones de gènes
d'ARNr 16S
454 Warnecke et al.,
2007 Métagénome intestinal du termite 71 Mbases 454 Qin et al., 2010 Catalogue de gènes du microbiome intestinal
humain 576,7 Gbases Illumina GA et Sanger Le consortium sur le projet du microbiome humain, 2012a Fonction et diversité du microbiome humain chez
le sujet sain 2Tbases Illumina GA et 454 Le consortium sur le
projet du
microbiome humain, 2012b
Un cadre de travail pour la recherche sur le
microbiome humain 3,5Tbases
Illumina GA Abubucker et al., 2012 Reconstructions métaboliques pour le
microbiome humain 3,5Tbases
Illumina GA
Dans les dernières années, on a pu constater l'émergence de nouvelles études métagénomiques basées sur l'expression des gènes (Urich et al., 2008; Moran, 2009) ou sur la production de protéines (Wilmes et al., 2008; Schweder et al., 2008) plutôt que sur l'ADN. En effet, la métatranscriptomique et la métaprotéomique (voir section 2.4.1.) sont des approches qui ont le potentiel de nous apporter de nouvelles connaissances sur la dynamique fonctionnelle des communautés microbiennes (Simon & Daniel, 2011).
2.1.4. Défis
Conceptuellement, une approche métagénomique semble plutôt simple (Figure 2.1.); il suffit d'extraire, puis de séquencer les acides nucléiques d'un échantillon environnemental, pour ensuite analyser le tout afin de caractériser la communauté microbienne de l'échantillon en question. Mais en réalité, l'utilisation de la métagénomique amène certains défis et limites dont il faut tenir compte lors de la conception de la méthodologie d'un projet et dans l'interprétation des résultats obtenus. Les éventuels problèmes associés à une étude de métagénomique peuvent être répertoriés selon les aspects détaillés dans les sous-sections 2.1.4.1. à 2.1.4.4.
2.1.4.1. L'environnement analysé
Il faut toujours être conscient que l'échantillonnage d'un environnement donné se fait en un point géographique bien particulier, et à un moment précis dans le temps, et que par conséquent, la composition d'une communauté peut varier grandement si l'un ou l'autre de ces paramètres change (Morris et al., 2002; Kunin et al., 2008).
De plus, la notion de microenvironnement vient compliquer le processus d'échantillonnage, qui généralement n'est pas assez précis pour différencier les sous-écosystèmes qui peuvent coexister dans un même environnement. Un gramme de sol forestier par exemple, peut contenir de 6400 à 38000 espèces différentes de bactéries (Ederer, 2011), cependant, ces dernières ne sont pas distribuées de façon égale dans le sol, il est bien plus probable qu'elles forment des mini-communautés organisées en sous-écosystèmes pouvant être en contact ou séparés les uns des autres. Ainsi, l'isolement de l'ADN d'un échantillon contenant ce type de microenvironnement ne permettra vraisemblablement pas de reconstituer la sous-distribution des membres qui composent ce genre de mini-communautés.
D'autre part, le principe même de la métagénomique fait en sorte que les fragments séquencés à partir d'un échantillon d'ADN génomique sont issus d'un plus ou moins grand nombre d'espèces différentes dont, pour la majeure partie, le génome complet n'est pas présent. L'identification de l'espèce d'origine auquel appartient un fragment donné n'est donc pas chose aisée. Nous traitons ce sujet plus en détail dans la section 2.3.3. de cette thèse.
Une étude de Huber et collaborateurs (Huber et al., 2007) sur des communautés de bactéries et d'archées vivant dans des cheminées hydrothermales, a montré qu'un échantillon métagénomique peut être représentatif de l'environnement en termes de taxonomie (en l'occurrence de la population d'archées dans cet exemple). Cependant, ces mêmes auteurs ne sont pas parvenus à explorer de façon exhaustive la population bactérienne de cet environnement, cette dernière étant beaucoup plus diversifiée. En effet, la difficulté d'identifier potentiellement toutes les espèces d'un écosystème augmente avec la complexité et diversité taxonomique de ce dernier (Sogin et al., 2006; Huber et al., 2007; Wooley et al., 2010). Même un séquençage « en profondeur » d’un environnement naturel permet d’accéder seulement à une fraction infime de la variabilité génétique disponible dans le milieu en question (Gilbert & Dupont, 2011). Par conséquent, une analyse
métagénomique permet habituellement d'identifier seulement les membres les plus abondants d'une communauté (Tyson et al., 2004), car les espèces les plus nombreuses masquent celles qui sont rares, en fournissant une part beaucoup plus importante du matériel génétique total de la population microbienne en question. Ainsi, les chances de retrouver des fragments appartenant aux espèces moins représentées dans l'échantillon sont plus faibles. À titre d’exemple, une étude sur un échantillon d’une communauté microbienne de rumen comptant plus de 500 genres bactériens différents, a mis en évidence que les séquences issues des cinq plus abondants genres comptaient pour plus de 73% du nombre de séquences totales (Sparks et al., 2011). Néanmoins, certaines techniques comme l'hybridation différentielle permettent de soustraire l'ADN le plus abondant d'un échantillon afin de permettre d'avoir accès aux génomes des espèces plus rares (Galbraith et al., 2007). D'une manière générale, il faut donc être prudent quant à la représentativité des résultats obtenus lors d'une étude métagénomique. En d'autres termes, ce qui est observé dans l'échantillon ne le sera pas forcément dans l'environnement dont ce dernier est issu. D'autant plus que Bent et Forney (Bent & Forney, 2008) ont mis en évidence que la majorité des communautés microbiennes complexes affichent une distribution taxonomique caractéristique où seulement quelques espèces dominent alors que la vaste majorité de la population est inusitée.
2.1.4.2. L'étape d'isolation et d'extraction des acides nucléiques
Les microorganismes ont différents niveaux d'accessibilité face à l'isolation. Certains d'entre eux peuvent rester attachés à des particules inertes présentes dans l'échantillon, rendant leur ADN inaccessible. De plus, l'efficacité du procédé de lyse cellulaire varie selon les organismes, si bien que le produit d'extraction d'un échantillon environnemental ne représentera pas forcément tous les organismes présents au départ lors de l'échantillonnage. Ceci pouvant bien évidemment biaiser l'estimation de la composition taxonomique de l'échantillon en question.
Il a été rapporté récemment que l'utilisation de différentes approches d'extraction pour l'étude d'un même métagénome apporte l'avantage de pouvoir caractériser des parties différentes de ce dernier, mais aussi d'augmenter le nombre d'espèces et de fonctions détectées (Delmont et al., 2012). Cependant, Morgan et collaborateurs (Morgan et al.,
2010) ont démontré que l'utilisation de protocoles d'extraction différents dans des études de métagénomique comparative peuvent entraîner des biais significatifs dans les résultats; c'est la raison pour laquelle nous avons choisi d'avoir recours à la même méthode d'extraction pour tous les échantillons analysés dans nos études, nous permettant ainsi de considérer les mêmes biais dans toutes nos analyses subséquentes.
2.1.4.3. L'étape de clonage
La fabrication d'une librairie de clones préalablement au séquençage peut entraîner certains biais inhérents au processus de clonage (niveau d'efficacité, nombres suffisants de clones produits), mais aussi à l'étape de PCR qui est souvent nécessaire après un clonage, et qui comporte certains désavantages comme les biais introduits par l'utilisation des sondes et/ou par l'amplification exponentielle (Suzuki & Giovannoni, 1996; Wang & Wang, 1997; Kanagawa, 2003; Acinas et al., 2005; Sipos et al., 2010). On sait par ailleurs que des fragments d'ADN pauvres en bases GC peuvent être moins stables et affecter l'efficacité du clonage (Temperton et al., 2009). De plus, certains gènes ne peuvent être incorporés dans les vecteurs de librairie en raison de leur instabilité ou de leur toxicité pour la cellule hôte (Sorek et al., 2007).
Néanmoins, l'ensemble de ces biais peuvent être évités en supprimant tout simplement l'étape de clonage, et donc en séquençant directement l'ADN génomique obtenu après extraction. Cette approche a d'ailleurs été proposée comme la méthode la plus précise pour l'estimation de la diversité taxonomique de métagénomes (von Mering et al., 2007).
2.1.4.4. Le séquençage
La présence de certaines substances inhibitrices contenues dans l'environnement prélevé, comme l'acide humique souvent associée aux échantillons provenant du sol, peut affecter l'efficacité du séquençage.
Il faut mentionner également qu'il existe d'autres défis liés à l'utilisation de la métagénomique, mais étant donné que ceux-ci sont associés au séquençage à haut débit et au traitement bio-informatique des séquences générées, j'ai fait le choix de les aborder dans les sections 2.2. et 2.3. respectivement.
2.2. Séquençage à haut débit
L'essor de la métagénomique n'aurait pu avoir lieu sans les remarquables avancées technologiques que le séquençage de l'ADN a connu lors de la dernière décennie, notamment avec l'avènement des techniques de séquençage de deuxième génération, qui permettent de produire un nombre beaucoup plus important d'ADN séquencés, en moins de temps et à moindre coût.
2.2.1. Historique
Au cours du 20ème siècle, le séquençage de l'ADN a révolutionné la recherche dans le domaine des sciences de la vie, en réalisant un grand pas pour l'humanité avec le séquençage complet du génome humain en 2001 (Lander et al., 2001; Venter et al., 2001). Historiquement, ceci a été rendu possible grâce aux prouesses de deux pionniers travaillant sur le séquençage, W. Gilbert et F. Sanger qui se partagèrent le prix Nobel de chimie en 1980 pour leurs co-découvertes dans ce domaine. Tout a commencé en 1975 avec Sanger et Coulson qui sont parvenus à développer la méthode de terminaison des chaînes, permettant le séquençage d'environ 50 nucléotides contigus sur plusieurs jours (Sanger & Coulson, 1975). Deux ans plus tard, Maxam et Gilbert présentent alors une autre méthode de séquençage basée sur la dégradation chimique de l'ADN par coupures sélectives (Maxam & Gilbert, 1977). Cependant, la même année Sanger publie une méthode complémentaire à celle qu'il avait déjà créé en 1975, en introduisant l'utilisation des didéoxynucléotides, des inhibiteurs de l'ADN polymérase spécifiques à chaque base nucléotidique (Sanger et al., 1977). La même année, le développement de cette technologie a ainsi permis de séquencer le tout premier génome complet, celui du phage φX (Sanger et al., 1977). Dans les deux décennies suivantes, la méthode de séquençage de Sanger a été améliorée en termes de procédés et de chimie, avec la capacité de produire des séquences d'une longueur de 1000 nucléotides avec une précision pouvant atteindre 99,999% (Shendure et al., 2004). Mais l'avancée majeure depuis cette optimisation reste son automatisation, qui a ouvert la voie à la démocratisation du séquençage à partir des années 1990 (Hutchison, 2007). Ces avancées dans la technique de Sanger ont rendu possible le séquençage d'une variété de génomes bactériens et eucaryotes, dont les plus marquants sont : Saccharomyces cerevisiae (Goffeau et al., 1996), Escherichia coli (Blattner et al., 1997), le riz (Goff et al., 2002; Yu et al.,
2002), la souris (Waterston et al., 2002), et enfin l'homme (Lander et al., 2001; Venter et al., 2001).
Néanmoins, la démocratisation du séquençage en aveugle de génomes dans les années 1990, a conduit à la génération d'un volume de plus en plus important de données à séquencer, rendant ainsi la technologie de Sanger souvent inadéquate, car jugée trop longue et particulièrement onéreuse (Shendure & Ji, 2008). C'est la raison pour laquelle en 2004, le National Human Research Institute a lancé « le programme de séquençage du génome à 1000$ » dans le but de réduire à cette somme le prix du séquençage des génomes de mammifère, et ceci d'ici une dizaine d'années. Ainsi après cette annonce, plusieurs compagnies se sont lancées dans l'aventure afin de créer des nouveaux instruments de séquençage permettant de produire plus rapidement, et pour moins cher, des quantités massives de données (Meldrum, 2001; Meldrum & Holl, 2002). C'est dans cet esprit que sont nées les technologies de séquençage à haut débit de deuxième et troisième génération.
2.2.2. Le séquençage en aveugle
Ce type de séquençage (appelé « whole-genome shotgun sequencing ») a été popularisé à la fin des années 1990 par l'Institut Craig Venter, bien que Sanger et collaborateurs l'avaient proposé dès 1982 avec le séquençage du génome complet du phage λ (Sanger et al., 1982). Cette méthode, toujours utilisée à l'heure actuelle, a permis l'assemblage de grands génomes (bactériens puis eucaryotes) tels que ceux cités dans la section 2.2.1.
1. L'ADN génomique de l'organisme à séquencer est fragmenté de façon aléatoire;
2. Les fragments sont clonés au hasard dans un vecteur (plasmide ou BAC), qui est ensuite introduit dans une bactérie hôte (comme E. coli) afin de créer une librairie de séquences et de produire ainsi assez de matériel pour le séquençage subséquent;
3. Les fragments de la librairie de clones sont alors séquencés avec la technologie de Sanger;
4. Les séquences obtenues sont assemblées à l'aide de différents outils de bio-informatique afin de reconstituer le génome complet de l'organisme en question.
Signalons cependant, que ce type de séquençage est de plus en plus concurrencé par l'avènement des technologies de séquençage dites de « nouvelle génération » présentées dans la section suivante.
2.2.3. Les méthodes à haut débit
Apparues à partir de 2005, les méthodes de séquençage à haut débit de l'ADN, dites de « nouvelle génération » (Next Generation Sequencing (NGS)), ont gagné beaucoup de terrain sur la technologie de Sanger pour le séquençage des petits génomes et des écosystèmes environnementaux. Ceci est principalement dû au fait qu’elles sont grandement parallélisées permettant la production de plusieurs millions de séquences (« reads ») différentes par expérience sans avoir nécessairement besoin d’une étape de clonage au préalable. Ainsi, ces méthodes permettent d’obtenir un plus grand nombre de séquences, plus rapidement et pour un prix inférieur à la méthode de Sanger. Néanmoins, les « reads » produits sont plus courts pour la plupart des technologies existantes en 2012: de 35 à 250 pb (paires de bases), excepté pour le pyroséquençage 454 qui depuis peu, serait en mesure de séquencer des fragments d'une longueur moyenne de 700 pb égalant ainsi plus ou moins la méthode de Sanger (Siqueira et al., 2012).