Développement d’une thérapie pour
l’Ataxie de Friedriech basée sur l’administration des
protéines Tat-Frataxine et Pep-1-Frataxine
Mémoire
Amina Chikh
Maîtrise en biologie cellulaire et moléculaire
Maître ès sciences (M.Sc.)
Québec, Canada
iii
L’Ataxie de Friedreich est une maladie génétique rare et grave, associant une neurodégénérescence, une cardiomyopathie et un diabète. Elle est causée par une réduction drastique d’une protéine mitochondriale appelée frataxine. L’approche de notre laboratoire sera donc de développer sur des cellules en culture et dans un modèle animal une thérapie moléculaire qui aura pour but de fournir aux cellules une protéine frataxine recombinante et réduire si possible les symptômes de la maladie. Afin de permettre la transduction de la frataxine, nous l'avons fusionné à un domaine de transduction protéique (Cell Penetrating Peptides, CPP) comme Tat ou Pep-1. Ces peptides sont bien connus pour leur capacité d’acheminer, dans les cellules, les protéines auxquelles ils sont fusionnés par des vésicules d’endocytose, mais également d’être libérées de ces derniers pour participer au métabolisme de la cellule. Des observations préliminaires obtenues, nous concluons que les protéines Tat-Frataxine et Pep-1-Frataxine assurent in vitro et in vivo la viabilité des cellules déficientes en frataxine endogène.
v
ABSTRACT
Friedreich Ataxia is a rare and serious genetic disease involving neurodegeneration, cardiomyopathy and diabetes. It is caused by a drastic reduction of a mitochondrial protein called frataxin. The approach of the laboratory will be to develop on in vitro cells and in vivo mice models, a molecular therapy that will aim to provide the cells with a recombinant protein frataxin and if possible reduce the symptoms of the disease. To enable transduction of frataxin, we fused it to a protein transduction domain (Cell Penetrating Peptides, CPP), Tat or Pep-1. These peptides are well known for their ability to allow the penetration into the cells of the proteins to which they are fused through endocytosis vesicles, but also to be released from these vesicles to take part in the cell metabolism. Preliminary observations led us to conclude that the Tat-Frataxin and Pep-1-Frataxin protein enhance
vii
RÉSUMÉ ... iii
ABSTRACT ... v
TABLE DES MATIÈRES ... vii
LISTE DES FIGURES ... xi
LISTE DES TABLEAUX ... xiii
LISTE DES ABRÉVIATIONS ... xv
INTRODUCTION ... 1
CHAPITRE I : L’ATAXIE DE FRIEDREICH ... 3
1. Les ataxies ... 3 2. L’Ataxie de Friedriech ... 5 2.1. Historique ... 5 2.2. Epidémiologie ... 6 3. Caractéristiques cliniques ... 6 3.1. Syndrome neurologique ... 7 3.2. Syndrome ostéo-articulaire ... 7
3.3. Syndrome viscéral et endocrinien ... 9
3.3.1. Atteinte cardiaque ... 9
3.3.2. Diabète ... 9
4. Caractéristiques anatomopathologiques ... 11
4.1. Au niveau du système nerveux central ... 11
4.2. Au niveau du système nerveux périphérique ... 11
5. Caractéristiques étiologiques ... 13
5.1. L’Ataxie de Friedreich: Au niveau moléculaire ... 13
5.1.1. Le gène responsable de l’Ataxie de Friedreich ... 13
5.1.2. Les mutations associées à l’Ataxie de Friedreich ... 13
5.2. L’Ataxie de Friedreich : Au niveau cellulaire ... 16
5.2.1. Structure de la protéine frataxine ... 16
5.2.2. Localisation et maturation mitochondriale de la frataxine ... 16
viii
6. Modèles murins ... 19
6.1. Souris Frda / MCK et Frda / NSE ... 19
6.2. Souris YG8R ... 19
7. Les approches thérapeutiques ... 20
7.1. Diagnostic prénatal ... 20
7.2. Thérapie pharmacologique ... 20
7.3. Thérapie génique et autres perspectives ... 21
CHAPITRE II : LES PEPTIDES DE PÉNÉTRATION CELLULAIRE ... 23
1. Généralité ... 23
2. Mécanismes d’entrée dans la cellule ... 26
2.1. La translocation directe ... 26
2.2. L’endocytose ... 26
3. Exemples de CPP ... 29
3.1. Le peptide Tat ... 29
3.2. Le peptide Pep-1 ... 30
CHAPITRE III : HYPOTHÈSE DE TRAVAIL ET OBJECTIFS ... 31
1. Hypothèse de travail ... 31
2. Objectifs ... 31
1. Production des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 ... 33
1.1. Clonage et construction des vecteurs d’expression bactériens ... 33
1.1.1. Le système d’expression pET chez Escherichia coli ... 33
1.1.2. Amplification du gène FXN ... 34
1.1.3. Sous-clonage dans le vecteur pDrive ... 35
1.1.4. Construction des vecteurs pET-16b-6xHis-Tat-Fxn et pET-16b-6His-Pep-1-Fxn ... 36
1.1.5. Addition de l’épitope V5 ... 36
1.2. Transformation des bactéries compétentes BL21 ... 38
1.3. Expression des protéines recombinantes ... 38
1.3.1. Culture des bactéries transformées ... 38
1.3.2. Induction par l’IPTG ... 38
1.4. Extraction et purification des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 ... 39
ix
1.4.1. Lyse des cellules bactériennes ... 39
1.4.2. Purification des protéines recombinantes en conditions natives ... 39
1.4.3. Dialyse des protéines recombinantes ... 39
2. Évaluations biologiques des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 in vitro et in vivo ... 40
2.1. Essais in vitro des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 40 2.1.1. Transduction des protéines recombinantes dans les lymphoblastes humains ... 40
2.1.1.1. Culture des lymphoblastes humains ... 40
2.1.1.2. Transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5………40
2.1.1.3. Détermination de l’activité métabolique par le test XTT ... 41
2.1.2. Transduction des protéines 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 dans les fibroblastes murins ... 41
2.1.2.1. Culture des fibroblastes murins KO / conditionnel KO ... 41
2.1.2.2. Transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5………42
2.1.3. Analyse par immunobuvardage de type Western ... 45
2.1.3.1. Détection de la protéine Cre recombinase ... 45
2.1.3.2. Détection des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5………46
2.2. Essais in vivo des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 47 2.2.1. Animaux ... 47
2.2.2. Injection par voie intra-péritonéale des protéines recombinantes et prélèvement d’organes………47
2.2.3. Quantification par le test ELISA ... 48
1. Résultats ... 51
1.1. Production des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 ... 51
1.1.1. Clonage et construction des vecteurs d’expression bactériens ... 51
1.1.2. Expression et purification des protéines recombinantes ... 51
1.2. Tests d’activité in vitro ... 54
1.2.1. Transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 dans les lymphoblastes humains ... 54
x
1.2.2. Transduction des protéines recombinantes dans les fibroblastes murins KO / conditionnel KO ... 56 1.3. Tests d’activité in vivo ... 65
1.3.2. Détection des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5
par le test ELISA ... 65 2. Discussion ... 67
2.1. Production des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 ... 68
2.2. Les protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 restaurent
l’activité métabolique des lymphoblastes FRDA... 68 2.3. Les protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 permettent la survie des fibroblastes murins totalement déficients en frataxine endogène... 69
2.4. L’administration par voie intra-péritonéale des protéines recombinantes
6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-6xHis-Tat-Fxn-V5 permet leur détection dans différents organes ... 70 CONCLUSION ... 71 ANNEXES ... 73 ANNEXE I : UNE NOUVELLE APPROCHE THÉRAPEUTIQUE POTENTIELLE POUR L’ATAXIE DE FRIEDREICH : INDUCTION DE L’EXPRESSION DE LA FRATXINE AVEC LES PROTÉINES TALE ... 75
ANNEXE II : Le AAV9 CODANT POUR LA FRATAXINE AMÉLIORE NETTEMENT LES SYMPTÔMES ET PROLOGE LA DURÉE DE VIE DES SOURIS MODÈLES DE L’ATAXIE DE FRIEDRIECH ... 77 BIBLIOGRAPHIE ... 79
xi
LISTE DES FIGURES
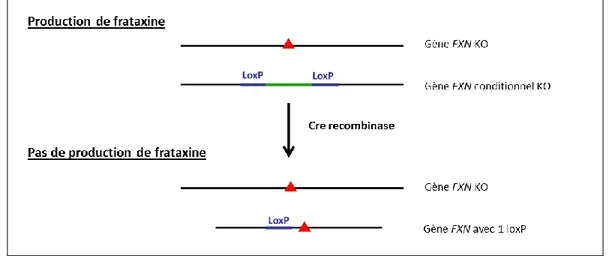
Figure 1 : Le pied creux bilatéral dans FRDA ... 8 Figure 2 : Hypertrophie cardiaque dans FRDA ... 10 Figure 3 : Les différents sites de dégénérescence au niveau du système nerveux central (SNC) et au niveau du système nerveux périphérique (SNP) ... 12 Figure 4 : Représentation schématique des mutations associées à l'Ataxie de Friedreich ... 15 Figure 5 : Schéma représentant les mécanismes de la chaine respiratoire et de la synthèse d’ATP par phosphorylation oxydative ... 18 Figure 6 : Les mécanismes d’entrée dans la cellule des peptides de pénétration cellulaire ... 28 Figure 7 : Carte du vecteur d’expression bactérien pET-16b ... 33 Figure 8 : Inactivation conditionnelle spatio-temporelle par le système Cre-LoxP du gène FXN dans des fibroblastes murins KO / conditionnel KO ... 42 Figure 9 : Expérience a. Inactivation conditionnelle spatio-temporelle (système Cre-LoxP) ... 43 Figure 10 : Expérience b. Transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et
6xHis-Pep-1-Fxn-V5 ... 44 Figure 11 : Expérience c. Inactivation conditionnelle spatio-temporelle (système Cre-LoxP) et transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 ... 44 Figure 12 : Schéma représentant l’immuno-dosage à flux latéral de type ELISA. ... 49 Figure 13 : Électrophorèse SDS-PAGE de la production des protéines recombinantes
6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-6xHis-Tat-Fxn-V5 par induction à l’IPTG chez E. coli ... 52 Figure 14 : Purification de la protéine recombinante 6xHis-Tat-Fxn-V5 (en condition natives) ... 53 Figure 15 : Purification de la protéine recombinante 6xHis-Pep-1-Fxn-V5 (en conditions natives) .... 53 Figure 16 : Comparaison de l’effet des protéines recombinantes 6xHis-Tat-Fxn-V5 et
6xHis-Pep-1-Fxn-V5 sur l’activité métabolique des lymphoblastes normaux et des lymphoblastes FRDA .... 55 Figure 17 : Inactivation conditionnelle spatio-temporelle (système Cre-LoxP) du gène FXN dans les fibroblastes murins KO / conditionnel KO ... 57 Figure 18 : Détection de la Cre recombinase dans les fibroblastes murins KO / conditionnel KO par immunobuvardage de type Western ... 58 Figure 19 : Transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 dans les fibroblastes murins KO / conditionnel KO ... 59
xii
Figure 20 : Détection des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 dans les fibroblastes murins KO / conditionnel KO ... 61 Figure 21 : Inactivation conditionnelle spatio-temporelle (système Cre-LoxP) et transduction des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 dans les fibroblastes murins KO / conditionnel KO ... 63 Figure 22 : Détection de la Cre recombinase et des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 dans les fibroblastes murins KO / conditionnel KO par immunobuvardage de type Western ... 64 Figure 23 : Détection des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 par le test ELISA. ... 66
xiii
LISTE DES TABLEAUX
Tableau 1 : Classification des ataxies récessives dégénératives proposée par Koenig (2003) ... 4
Tableau 2 : Les essais cliniques réalisés avec l’idébénone chez les patients FRDA ... 22
Tableau 3 : Les propriétés de quelques peptides de pénétration cellulaire (CPPs) ... 25
Tableau 4 : Amorces et conditions de l’amplification PCR de l'ADNc de la frataxine ... 34
Tableau 5 : Séquence d’acides aminés des protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5 ... 37
xv
LISTE DES ABRÉVIATIONS
AAV Adeno-associated virus
ADN Acide désoxyribonucléique
ADNc Acide désoxyribonucléique complémentaire
ADP Adénosine diphosphate
ARN Acide ribonucléique
ARNm Acide ribonucléique messager
ATP Adénosine triphosphate
BCA Acide bicinchonique
BHE Barrière hémato-encéphalique
BSA Bovine serum albumin
CCPA Conseil canadien de protection des animaux
COX Cytochrome c oxidase
CPP Cell penetrating peptide
Cre Cycling recombination
DMEM Dulbecco's modified Eagle medium
DTT Dithiothreitol
eGFP Enhanced Green Fluorescent Protein
FBS Fetal bovin serum
FRDA Friedreich Ataxia
FXN Frataxine
g Force relative de centrifugation
GAA Guanine-Adénine-Adénine
Gln Glutamine
HBSS Hanks balanced salt solution
HRP Horseradish peroxidase
xvi IPTG Isopropyl-β-thiogalactopyranoside Kb Kilobase kDa KiloDalton KO Knock-out LB Luria-Bertani Lys Lysine
MCK Muscle creatine kinase
MCS Multiple clonage site
ml Millilitre
mM Millimolaire
MPP Mitochondrial processing peptidase
NaCl Chlorure de sodium
NPI Sodium phosphate imidazole (tampon)
NSE Neuron specific enolase
pb Paire de bases
PCR Polymerase chain reaction
Pen Pénétratine
PMS Méthosulfate de phénazine
Pro Proline
P/S Pénicilline / streptomycine
PTD Protein transduction domain
rpm Rotations par minute
SDS Sodium dodécyl sulfate
SDS-PAGE Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis
Ser Sérine
siARN Small interfering ARN
xvii
SNC Système nerveux central
SNP Système nerveux périphérique
TALE Transcription activator-like effector
TAR Trans-activator responsive element
Tat Trans-activateur de transcription
xix À mes parents, sans qui je n’en serais pas là aujourd’hui…,
xxi
Je tiens à exprimer mes sincères remerciements à mon directeur de recherche le Dr. Jacques-P.Tremblay. Ses encouragements, ses conseils ainsi que sa gentillesse m'ont spécialement aidé durant toute la période de ma formation. Qu’il trouve dans ces lignes mon profond respect et toute ma reconnaissance.
1
De nombreuses maladies neurodégénératives telles que l’Ataxie de Friedreich, la maladie de Parkinson et la maladie d’Alzheimer sont dues à des perturbations dans le fonctionnement mitochondrial et aux dommages oxydatifs. L’Ataxie de Friedreich (FRDA) constitue la forme la plus courante des ataxies héréditaires humaines, affecte particulièrement les populations d’origine européenne et touche une personne sur 50 000 (Lopez-Arlandis, Vilchez et al. 1995, Cossee, Campuzano et al. 1997). C’est une maladie neurologique sévère à transmission autosomique récessive, caractérisée par une dégénérescence spinocérébelleuse, à savoir une atteinte des voies allant du cervelet à la moelle épinière. Les personnes touchées souffrent de problèmes de coordination des mouvements volontaires (autrement dit: ataxie). Des signes extra-neurologiques sont aussi présents telles une cardiomyopathie hypertrophique et une incidence accrue de diabète. L’apparition des premiers symptômes est progressive et l’âge de début de la maladie se situe généralement entre 5 et 25 ans.
La mutation majoritaire associée à FRDA est une expansion homozygote d'une séquence répétée de triplets Guanine-Adénine-Adénine (GAA) au niveau du 1er intron du gène FXN (Campuzano,
Montermini et al. 1996). Les expansions trinucléotidiques sont des mutations dynamiques qui présentent une instabilité (augmentation / diminution) dans les cellules germinales mais également dans certaines cellules somatiques. Dans FRDA, l’expansion GAA est à l’origine d’une diminution partielle du taux d’expression du gène FXN et de la protéine impliquée dans FRDA, la frataxine
(Campuzano, Montermini et al. 1996, Cossee, Campuzano et al. 1997). Le nombre de répétition GAA peut aller de 150 à 1300 répétitions (ce triplet est normalement répété entre 7 et 35 fois) et cette expansion est inversement corrélée avec l’âge de début et la sévérité de la maladie (Durr, Cossee et al. 1996, Lamont, Davis et al. 1997).
La fonction précise de la frataxine n’est pas encore bien élucidée, néanmoins, sa réduction se traduit par l’agrégation des centres fer-soufre, d’une perturbation de l’homéostasie du fer et le maintien du potentiel d’oxydo-réduction (Santos, Lefevre et al. 2010).
Il n’existe actuellement aucun traitement curatif pour FRDA. Les thérapeutiques développées visent principalement deux voies: le traitement des conséquences métaboliques à l’origine du déficit en frataxine (idébénone, CoQ10 + vitamine E, pioglitazone, deferiprone) et l’augmentation des
2
concentrations cellulaires en frataxine (érythropoïétine, Histone déacétylase) (Santos, Lefevre et al. 2010). Ces molécules administrées aux patients permettent au mieux des soins palliatifs.
L’approche de notre laboratoire vise à augmenter le taux en frataxine retrouvé chez les patients FRDA par l’administration d’une protéine frataxine thérapeutique, la Tat- Fxn ou le Pep1-Fxn, dans le but de restituer toutes les fonctions essentielles de la frataxine chez ces patients. Notre travail comporte 3 étapes: (1) la construction des vecteurs plasmidiques 16b-6xHis-Tat-Fxn-V5 et pET-16b-6xHis-Pep-1-Fxn-V5 codant respectivement pour les protéines recombinantes 6xHis-Tat-Fxn-V5 et 6xHis-Pep-1-Fxn-V5, (2) suivie de la production et la purification de ces protéines frataxines recombinantes, et enfin (3), tester leurs activités biologiques in vitro (sur des lymphoblastes issus d’un patient FRDA et des fibroblastes murins ayant un gène FXN Knock-Out et un gène conditionnellement non-fonctionnel, Knock-Out) et in vivo (sur des souris Frda / MCK)).
À partir des résultats obtenus, nous concluons d’une part que les protéines Tat-Fxn-V5 et 6His-Pep-1-Fxn-V5 ont la capacité de transduire des cellules in vitro et in vivo et d’autre part, elles restaurent le défaut mitochondrial lié au déficit en frataxine endogène. Ainsi, il est envisageable de concevoir une approche thérapeutique pour FRDA basée sur l'administration systémique d'une protéine recombinante 6His-Tat-Fxn et 6His-Pep-1-Fxn. Les prochaines expériences devraient permettre de démontrer que notre approche est tout d’abord efficace dans un modèle murin adéquat tout en étant non immunogène.
3
CHAPITRE I : L’ATAXIE DE FRIEDREICH
1. Les ataxiesLe terme ataxie (du grec. a, sans; taxis, ordre) définit une pathologie neuromusculaire caractérisée par un manque de coordination des mouvements volontaires sans perte de la force musculaire
(Barboi 2000). Le trouble de la coordination des mouvements désigne la conséquence d’une atteinte des voies proprioceptives (fibres myélinisées de gros calibres du système nerveux périphérique, ganglions rachidiens postérieurs, colonnes postérieures de la moelle épinière, thalamus, cortex pariétal) mais également des lésions du cervelet et de ses connections afférentes et efférentes. L’ataxie peut se présenter de manière sporadique (traumatisme crânien ou privation d’oxygène à la naissance, tumeur au cerveau, maladie neurodégénérative, infection…) ou héréditaire par différents modes de transmission soit autosomique récessive, autosomique dominante (ataxies spinocérébelleuses et ataxies épisodiques), liée au chromosome X ou mitochondriale (Manto and Marmolino 2009).
Les ataxies héréditaires comprennent un groupe hétérogène de maladies neurodégénératives caractérisées par une dégénérescence du cervelet et / ou de la moelle épinière associée à une combinaison de signes neurologiques (neuropathie, épilepsie) et des signes extra-neurologiques (cardiomyopathie, le diabète, la surdité, la rétinite pigmentaire) (Taroni and DiDonato 2004). Un grand nombre de classifications ont été proposées pour les ataxies héréditaires qui différent en fonction des critères utilisés pour les classer. Ainsi, on distingue des classifications selon le mode de
transmission, les sites de neurodégénérescences ou les gènes associés à la pathologie (Tableau 1).
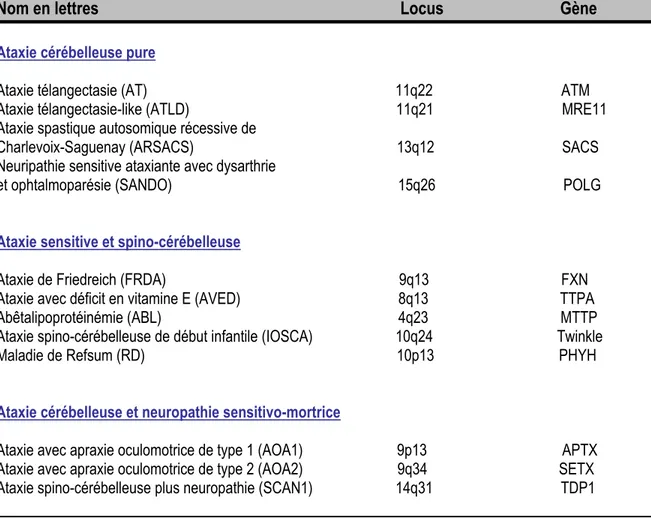
4
Tableau 1 :Classification des ataxies récessives dégénératives proposée par Koenig (2003)
(Tableau tiré de (Brahim 2009))
Nom en lettres Locus Gène
Ataxie cérébelleuse pure
Ataxie télangectasie (AT) 11q22 ATM Ataxie télangectasie-like (ATLD) 11q21 MRE11 Ataxie spastique autosomique récessive de
Charlevoix-Saguenay (ARSACS) 13q12 SACS Neuripathie sensitive ataxiante avec dysarthrie
et ophtalmoparésie (SANDO) 15q26 POLG
Ataxie sensitive et spino-cérébelleuse
Ataxie de Friedreich (FRDA) 9q13 FXN Ataxie avec déficit en vitamine E (AVED) 8q13 TTPA Abêtalipoprotéinémie (ABL) 4q23 MTTP Ataxie spino-cérébelleuse de début infantile (IOSCA) 10q24 Twinkle Maladie de Refsum (RD) 10p13 PHYH
Ataxie cérébelleuse et neuropathie sensitivo-mortrice
Ataxie avec apraxie oculomotrice de type 1 (AOA1) 9p13 APTX Ataxie avec apraxie oculomotrice de type 2 (AOA2) 9q34 SETX Ataxie spino-cérébelleuse plus neuropathie (SCAN1) 14q31 TDP1
5 2. L’Ataxie de Friedriech
L’Ataxie de Friedreich (FRDA) est une maladie neurologique fatale et est l’ataxie héréditaire la plus répandue à travers le monde. Un dysfonctionnement mitochondrial associé à des dommages oxydatifs sont à l’origine de cette maladie. Le mode de transmission est de type autosomique récessif caractérisé par une dégénérescence progressive du système nerveux central et du système nerveux périphérique accompagnée d’une cardiomyopathie hypertrophique et une prévalence élevée de diabète. Chez la majorité des patients FRDA, la mutation responsable est une expansion d'une répétition de triplet GAA au niveau du 1er intron du gène FXN, qui code pour une protéine mitochondriale, la frataxine. Les patients atteints présentent un déficit quantitatif en cette protéine, ce qui perturbe l’activité de la mitochondrie, un organite essentiel dans la cellule qui a pour rôle fondamental la production d’énergie. Un déficit énergétique qui touche particulièrement les tissus nerveux (cervelet et moelle épinière) et cardiaque, conduit à une insuffisance cardiaque fatale.
2.1. Historique
FDRA a été décrite pour la première fois par le neurologue allemand Nikolaus Friedreich. Il a rapporté les caractéristiques pathologiques et cliniques essentielles de la maladie à travers l’observation de neuf patients provenant de trois familles différentes. Il a remarqué que cette maladie était différente d’une ataxie ordinaire, ainsi il ressort à travers de ses observations chez ces patients, une ataxie progressive, perte sensorielle, une faiblesse musculaire des membres inférieurs suivie des membres supérieurs et une cardiomyopathie. Il a aussi mis en évidence le caractère familial de la maladie. Il a conclu qu’elle pouvait atteindre les deux sexes d’une même fratrie mais jamais les parents. Certains médecins attribuaient le diagnostic de la maladie chez ces patients à une forme de neurosyphilis ou d’un variant de la maladie de Charcot-Marie-Tooth de type I. Cependant, Nikolaus Friedreich a été en mesure de déterminer toutes les caractéristiques cliniques et pathologiques essentielles de la maladie, à l’exception de la perte des réflexes ostéo-tendineux, ceux-ci étant été décrits plus tard. La maladie a été nommée Ataxie de Friedreich (FRDA) en 1882 par P. Brousse. C’est à partir des années 1970 que des études ont été établies sur de grandes séries de patients afin de définir des critères de diagnostiques clairs. Le Groupe Collaboratif du Québec (Geoffroy et al. 1976) et Anita Harding (Harding 1981) avaient estimé que ces critères étaient trop stricts en raison
6
de certains symptômes majeurs qui apparaissent au début de la maladie et les symptômes secondaires qui eux peuvent être présents au cours de l’évolution de la maladie.
Le caractère héréditaire autosomique récessive a été suggéré dès le début des années 1960 mais a été confirmé que 10 ans plus tard. Ce n’est qu’en 1996, par clonage positionnel que le gène responsable de FRDA et la mutation majoritaire par expansion trinucléotidique GAA ont été identifiés par l’équipe de Michel Koenig en collaboration avec l’équipe de Massimo Pandolfo (Campuzano, Montermini et al. 1996). Ce gène a été baptisé FXN et code pour une protéine appelée frataxine, localisée dans les mitochondries. Sa fonction exacte reste inconnue à ce jour.
2.2. Epidémiologie
FRDA représente la moitié des ataxies de type cérébelleuse neurodégénérative à transmission autosomique récessive. Dans la population caucasienne, elle a une incidence de 1 pour 50 000 personnes (Campuzano, Montermini et al. 1996). Cependant, cette affection reste très rare dans la population finlandaise, d’Afrique noire et inexistante dans la population japonaise (Labuda, Labuda et al. 2000, Juvonen, Kulmala et al. 2002). Il n’y a donc pas une diffusion homogène de cette maladie au niveau mondiale.
3. Caractéristiques cliniques
Les manifestations cliniques de FRDA sont caractérisées par une combinaison de :
- Syndrome neurologique qui comprend un syndrome cérébelleux statique suivi d’un syndrome cérébelleux cinétique, un syndrome radiculo-cordonal postérieur associé à une atteinte de la sensibilité profonde, un syndrome pyramidal, une neuropathie axonale et une dysarthrie. D’autres signes neurologiques sont observés comme une atteinte auditive et une atrophie optique ;
- Syndrome ostéo-articulaire marqué par un pied creux bilatéral et une scoliose ;
- Syndrome viscéral et endocrinien caractérisé par une cardiomyopathie hypertrophique qui survient généralement quelques années après l’apparition des premiers signes neurologiques associée le plus souvent à un diabète.
7
L’évolution de FRDA est progressive et lente. Les patients perdent la marche quelques années après l’apparition des premiers symptômes et décèdent souvent vers l’âge de 40 ans suite à des complications cardio-pulmonaires.
3.1. Syndrome neurologique
Le syndrome neurologique comprend une ataxie des membres inférieurs qui s’étend aux membres supérieurs à composante cérébelleuse et cordonale postérieure. Elle se manifeste par des troubles d’équilibre en station debout (ataxie statique), l’équilibre devient incertain suivi de troubles de l’exécution de mouvement dans l’espace et dans le temps (ataxie cinétique), la démarche devient instable et titubante.
L’atteinte du tronc cérébral est caractérisée par une dysarthrie, la voix devient explosive et saccadée avec des difficultés à coordonner respiration et phonation. L’absence des réflexes ostéo-tendineux est due à une neuropathie axonale sensitive avec une dégénérescence des neurones de la colonne de Clarke et des colonnes postérieurs et de faisceaux pyramidaux et spinocérébelleux de la moelle épinière. Les patients FRDA sont également sujets à des troubles sensitifs et ophtalmiques. Cependant, les atteintes auditives restent rares (C.Strazielle 2002).
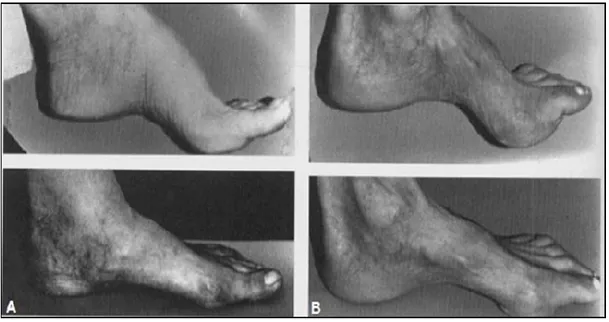
3.2. Syndrome ostéo-articulaire
La déformation du pied chez les patients FRDA correspond dans 75 % des cas par un pied creux bilatéral. Le pied creux est une déformation définie par une augmentation de la concavité plantaire se traduisant par un rapprochement des appuis plantaires antérieurs et postérieurs. Ainsi, la surface d’appui au sol s’en trouve diminuée (Figure 1). Cette déformation est de nature réductible donc flexible au début de la maladie mais progressivement le pied devient irréductible ne pouvant plus retrouver sa position normale. L’avant du pied présente quant à lui des déformations avec des orteils
en forme de griffe, accompagnées d’une hyperextension et hyperflexion des phalanges (Rüfenacht
2005).
La scoliose survient généralement à un stade avancé de la maladie (vers l’adolescence) avec une prévalence de 60 %. Elle est définie comme une déviation en «S» du rachis dans les 3 plans de l’espace, entrainant une torsion de la colonne vertébrale et des déformations du thorax ainsi que de l’abdomen. Les patients FRDA présentent une scoliose avec des courbures de plus de 10 degrés,
8
mais il n’est pas rare qu’ils présentent une cyphose thoracique ou thoraco-lombaire avec des courbures de 40 degrés ou plus. Même si la scoliose semble une déformation constante dans FRDA, elle n’est cependant pas toujours présente lorsque le diagnostic est établi (Dr Michel Rüfenacht 2004).
Figure 1 : Le pied creux bilatéral dans FRDA
A. Pied présentant une déformation réductible ; B. Pied présentant une déformation irréductible. (Photo tirée de (Rüfenacht 2005))
9 3.3. Syndrome viscéral et endocrinien
3.3.1. Atteinte cardiaque
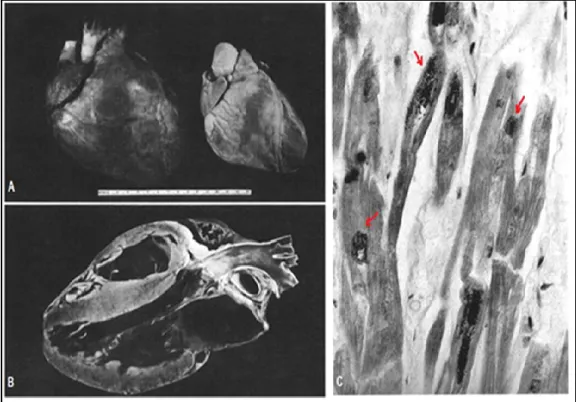
Les complications cardiaques apparaissent après les premiers symptômes neurologiques. Elles font partie des premières causes de décès des patients FRDA. Le cœur présente un épaississement des parois appelé « hypertrophie », observé au niveau du ventricule gauche avec une augmentation des parois et du septum qui est la cloison interventriculaire (Casazza, Ferrari et al. 1990, Kawai, Kato et al. 2000). Les quatre chambres cardiaques présentent également une dilation accompagnée parfois d’une hypertrophie des parois ventriculaires (Figure 2). De plus, des perturbations du rythme cardiaque ont été rapportées. La détection de l’hypertrophie cardiaque est établie par électrocardiographie et échocardiographie, ce qui permet de déceler les anomalies avant même l’apparition des symptômes car dans deux tiers des cas cette atteinte reste asymptomatique
(Pousset 2013).
3.3.2. Diabète
Le diabète se manifeste chez 10 à 20 % des patients FRDA. Bien que le rôle exact de la frataxine ne soit pas encore bien établi, un dysfonctionnement mitochondrial conduit généralement à un diabète dans certaines maladies. En effet, les mitochondries fournissent l’énergie nécessaire à la production de l’insuline par le pancréas. Dans le cas de FRDA, des études suggèrent un défaut de l’arginine dans l’induction de la sécrétion d’insuline, ce qui fait penser que le défaut génétique affecte
invariablement les cellules « bêta » des îlots de Langerhans du pancréas. Les études sont en cours afin de déterminer exactement le type de diabète dans FRDA (Cnop 2009), cependant jusqu'à présent la question reste sans réponse précise.
10
Figure 2 : Hypertrophie cardiaque dans FRDA
A. Un cœur présentant une hypertrophie cardiaque à gauche, en comparaison à droite avec un cœur normal. Ce sont deux cœurs de personnes de même âge ; B. Coupe dans un cœur hypertrophié montrant un épaississement de la paroi ventriculaire ; C. Coupe longitudinale d’un cœur hypertrophié visualisant le ventricule gauche avec accumulation de fer présentée en flèches rouges (coloration histologique de bleu de Prusse). (Photo tirée de (Simon 2004))
11 4. Caractéristiques anatomopathologiques
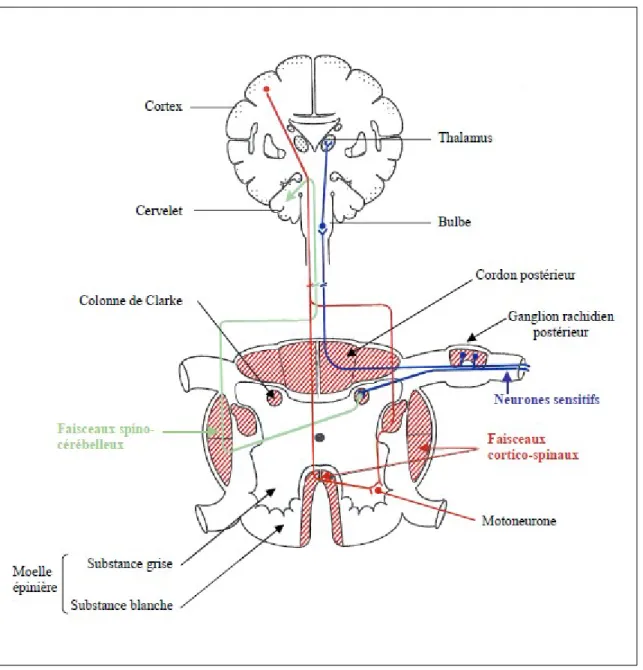
FRDA se distingue par une association de dégénérescence du système nerveux central et du système nerveux périphérique (cervelet, moelle épinière et ganglions de la racine dorsale) (Figure 3).
4.1. Au niveau du système nerveux central
Les altérations dégénératives touchent principalement des cordons postérieurs de la moelle épinière. Ces derniers prennent une apparence translucide et grisée. L’atrophie débute au niveau des ganglions rachidiens postérieurs, qui se traduit par une neuropathie sensitive axonale. Elle atteint ensuite, les cordons postérieurs de la moelle épinière, les faisceaux spinocérébelleux et les faisceaux pyramidaux qui sont le siège de la conduction des sensations proprioceptives des réflexes tendineux. De plus, une démyélinisation, une perte de fibres et une colonisation des cellules gliales sont observées au niveau des colonnes des Clarke et des tractus spinocérébelleux. Pour rappel, les neurones de la colonne de Clarke sont le siège des faisceaux spinocérébelleux et ont pour rôle de véhiculer les sensations proprioceptives inconscientes. Les faisceaux moteurs cortico-spinaux sont aussi atrophiés et cela se manifeste par un syndrome pyramidal.
Dans l’évolution de la maladie, les atteintes cérébelleuse et cérébrale apparaissent plus tardivement. Elles sont caractérisées par une perte neuronale qui se manifeste par une atrophie du noyau dentelé du cervelet et des voies cérébelleuses efférentes. La dégénérescence des voies spinocérébélleuse, des colonnes de Clarke et du noyau dentelé est à l’origine de l’ataxie cérébelleuse (Simon 2004).
4.2. Au niveau du système nerveux périphérique
Les lésions du système nerveux périphérique se caractérisent par une neuropathie axonale sensitive. Les informations sensitives transmises depuis la peau, des muscles et des articulations des membres sont véhiculées vers la moelle épinière par l’intermédiaire des voies afférentes. Les racines dorsales font remonter les informations aux noyaux sensitifs qui sont situés au niveau des ganglions rachidiens postérieurs. Dans FRDA, c’est une dégénérescence de fibres myélinisée de gros diamètre et une perte des corps cellulaires des neurones des ganglions rachidiens postérieurs. Cette dégénérescence est suivie par une prolifération des fibres de collagène ce qui démontre que la perte des fibres nerveuses se fait lentement et progressivement (Simon 2004).
12
Figure 3 : Les différents sites de dégénérescence au niveau du système nerveux central (SNC) et au
niveau du système nerveux périphérique (SNP)
13 5. Caractéristiques étiologiques
5.1. L’Ataxie de Friedreich: Au niveau moléculaire 5.1.1. Le gène responsable de l’Ataxie de Friedreich
Le gène responsable de FRDA a été identifié en 1996 par l’équipe du professeur Michel Koenig en collaboration avec l’équipe de Massimo Pandolfo. Ce gène a été baptisé «gène FXN ou X25», localisé sur le locus q13 du chromosome 9 et code pour la frataxine, une petite protéine de la
membrane mitochondriale (Campuzano, Montermini et al. 1996). Il comprend 7 exons (exon 1, exon
2, exon 3, exon 4, exon 5a, exon 5b et exon 6) et couvre une région génomique de 85 kb. Un épissage alternatif conduit à la formation de deux transcrits, un transcrit majoritaire comportant l’exon 5a (1,3 kb) codant pour la frataxine et un transcrit minoritaire qui contient les exons 5b et 6 (1,8 kb)
(Campuzano, Montermini et al. 1996, Cossee, Campuzano et al. 1997). Le gène FXN est exprimé de manière ubiquitaire dans les tissus humains, avec une fréquence plus importante au niveau du cœur, cerveau et moelle épinière comparativement au foie, au muscle squelettique et au pancréas
(Campuzano, Montermini et al. 1996, Koutnikova, Campuzano et al. 1997).
5.1.2. Les mutations associées à l’Ataxie de Friedreich
FRDA résulte dans la majorité des cas d’une expansion du nombre de copies de la répétition trinucléotidique GAA dans le 1er intron du gène FXN, plus précisément à 1,4 kb en aval de l’exon 1, au milieu d’une séquence Alu (GAA-Alu). Plus de 95 % des patients FRDA sont homozygotes pour cette expansion et le nombre de répétitions varie entre 150 à 1000 répétitions (la taille normale des répétitions GAA étant entre 7 et 35). Ce genre de mutation était une première dans une maladie génétique à transmission autosomique récessive (Campuzano, Montermini et al. 1996). Des mutations ponctuelles ont été décrites dans moins de 4 % des patients FRDA. Ces patients sont dits hétérozygotes composites, portant l’expansion GAA sur le 1er allèle et une mutation ponctuelle sur le 2ème allèle (Campuzano, Montermini et al. 1996, Cossee, Durr et al. 1999, De Castro, Garcia-Planells
et al. 2000). Parmi ces mutations ponctuelles, il a été répertorié 11 mutations tronquantes (regroupant des substitutions, des décalages du cadre de lecture par insertion ou délétion nucléotidique, des substitutions et des mutations non-sens) et 17 mutations faux-sens dues à une substitution d’un nucléotide (Figure 4).
14
L’'expansion GAA présentent une instabilité tant dans la transmission des parents / enfants que dans les différents tissus pendant le développement embryonnaire. En effet, la transmission de l’expansion GAA peut basculer soit vers une augmentation soit vers une diminution du nombre de répétitions d’une génération à une autre. Au niveau tissulaire, le nombre de répétions GAA varie d’un tissu à un autre et au sein du même tissu mais également tout au long du développement de l’individu
(Campuzano, Montermini et al. 1996).
Au niveau transcriptionnel, l’expansion GAA est à l’origine une diminution partielle du taux d’expression du gène FXN et de la frataxine par formation d’une structure d’ADN en triple hélice au niveau de cette expansion de façon proportionnelle avec le nombre de répétition GAA. Il pourrait s’agir également d’une condensation de la chromatine au niveau de l’expansion GAA formant ainsi
l’hétérochromatine en empêchant la progression de l’ARN polymérase (Ohshima, Montermini et al.
1998, Saveliev, Everett et al. 2003). L’âge de début de la maladie et la gravité des symptômes sont déterminés par la taille de l’expansion GAA. Ainsi, une corrélation inverse existe entre la diminution
de l’expression du gène FXN et la longueur de l’expansion GAA (Durr, Cossee et al. 1996, Cossee,
15 Figure 4 : Représentation schématique des mutations associées à l'Ataxie de Friedreich En flèche noire : l’expansion trinucléotidique (GAA) dans le 1er intron du gène FXN. Les mutations ponctuelles regroupent des substitutions au niveau du codon initiateur de la traduction ATG, des mutations des sites d’épissage (traits pointillés), des mutations faux-sens et non-sens (points rouges), des délétions (del) et des insertions (triangles verts). (Image tirée de (Simon 2004))
16
5.2. L’Ataxie de Friedreich : Au niveau cellulaire 5.2.1. Structure de la protéine frataxine
La frataxine est hautement conservée au cours de l’évolution depuis certaines bactéries Gram négatives jusqu’à l’Homme (Huynen, Snel et al. 2001). Elle se présente sous une structure globulaire monomérique compacte maintenue par un réseau hydrophobe. Elle est constituée principalement de deux régions distinctes: une région N-terminal qui correspond à une séquence d’adressage mitochondrial comprenant 15 résidus d’acides aminés et une région C-terminal composée d’une paire d’hélices α parallèles et de 7 feuillets β antiparallèles (Cho, Lee et al. 2000, Dhe-Paganon, Shigeta et al. 2000, Musco, Stier et al. 2000). Des résidus d’acides aminés sont localisés à la surface de la frataxine et forme ainsi la face hydrophobe et l’arête négative, ce qui lui confère la capacité d’interagir avec les protéines mitochondriales impliquées dans le métabolisme du fer (Adinolfi, Trifuoggi et al. 2002).
5.2.2. Localisation et maturation mitochondriale de la frataxine
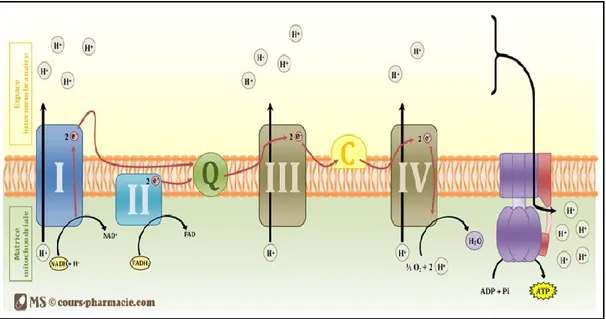
La frataxine est codée par le génome nucléaire et synthétisée sous forme de précurseur de 210 acides aminés, d’environ 25 kDa. Elle porte une séquence d’adressage mitochondriale qui conduit a proposé que la frataxine serait une protéine mitochondriale. La confirmation d’une localisation mitochondriale provient initialement d’études d’expression de la frataxine fusionnée à la β-galactosidase dans les cellules Hela qui ont permis de montrer sa localisation au niveau des membranes mitochondriales (Koutnikova, Campuzano et al. 1997). Actuellement, il est admis qu’il s’agit d’une protéine de la matrice mitochondriale (Babcock, de Silva et al. 1997, Long, Jirku et al. 2008).
Pour rappel, les mitochondries sont des organites qui jouent un rôle dans la production d’adénosine triphosphate (ATP) par phosphorylation oxydative au niveau de la chaîne respiratoire (Figure 5). La chaîne respiratoire est située dans la membrane interne des mitochondries, elle correspond à une association de complexes protéiques qui fonctionnent comme des transporteurs : le complexe I (NADH ubiquinone oxydoréductase), le complexe II (succinate ubiquinone oxydoréductase), le complexe III (l’ubiquinone-cytochrome c réductase) et le complexe IV (le cytochrome c oxydase ou COX) le complexe V (ATPase) qui a pour rôle la synthèse d’ATP à partir d’adénosine diphosphate (ADP) et du phosphate inorganique dans la matrice mitochondriale. Les mitochondries interviennent
17
également dans le métabolisme cellulaire en assurant le cycle de Krebs, la bêta-oxydation des acides gras et la synthèse de l’hème.
Le processus de maturation de la frataxine au niveau des mitochondries est réalisé par interaction entre une peptidase de clivage mitochondrial MPP (MPP, «Mitochondrial Processing Peptidase») et les acides aminés 4 à 87 de la partie N-terminal de la frataxine (Koutnikova 1998). C’est un clivage effectué en deux étapes successives, générant une forme intermédiaire lors du premier clivage entre les acides aminés 41 et 42(i-Fxn42-210: 19 kDa) suivi d’un deuxième clivage entre les résidus 80 et 81
donnant lieu à une protéine mature (m-Fxn81-210: 14.2 kDa) (Cavadini, Adamec et al. 2000, Condo,
Ventura et al. 2007).
5.2.3. Rôle de la frataxine
La fonction de la frataxine reste encore à déterminer de façon précise. Néanmoins, du fait de l’accumulation anormale de fer dans les mitochondries tant dans les modèles expérimentaux déficitaires en frataxine que dans le cœur des patients FRDA, la frataxine semble jouer un rôle clé dans le mécanisme de formation des centres Fe-S et le contrôle d'import et / ou d'export de fer à travers les membranes mitochondriales (Rotig, de Lonlay et al. 1997, Foury 1999). Les centres Fe-S sont essentiels dans le contrôle des transferts d’électron, la catalyse enzymatique, l’homéostasie du fer et la synthèse de l’hème. Dans la biogenèse des centres Fe-S, la frataxine contrôle l’entrée de fer au sein de la « scaffold » par activation de la cystéine désulfurase, l’asssemblage et la protection des centres Fe-S dans celle-ci (Colin, Martelli et al. 2013). La démonstration de l’implication de la frataxine comme source de fer et d’électrons, nécessaires dans le mécanisme de formation des centres Fe-S a été démontré dans le modèle de levure et les modèles de souris (Puccio, Simon et al. 2001, Muhlenhoff, Gerber et al. 2003).
Par ailleurs, des études complémentaires sur des cellules humaines ont par la suite confirmé son implication dans la régulation de la production et la maturation des centres Fe-S mitochondriale et cytosoliques (Stehling, Elsasser et al. 2004, Martelli, Napierala et al. 2012).
18
Figure 5 : Schéma représentant les mécanismes de la chaine respiratoire et de la synthèse d’ATP
par phosphorylation oxydative
La chaine respiratoire correspond à une chaine de complexes protéiques présents au sein de la membrane interne de la mitochondrie. Elle est responsable de la production d’ATP à partir du NADH et du FADH2 produits lors des différents voies catalytiques de l’organisme.
- Le complexe I a une action NADH coenzyme Q réductase, récupérant les électrons du NADH et
permet le transport de 4 protons de la matrice mitochondriale à l’espace inter-membranaire
- Le complexe II a une action Succinate coenzyme Q réductase, récupérant les électrons FADH2 et
ne permet pas le transport de proton
- Le complexe III a une action Coenzyme Q cytochrome C réductase, et permet le transport de 4
protons de la matrice mitochondriale à l’espace inter-membranaire
- Le complexe IV a une action Cytochrome C oxydase, et permet le transport de 2 protons de la
matrice mitochondriale à l’espace inter-membranaire
- Le coenzyme Q (ou ubiquinone) permet la transition entre le complexe I ou II et le complexe III - Le cytochrome C permet la transition entre le complexe III et le complexe IV
19 6. Modèles murins
Il est indispensable de générer des modèles animaux afin de mieux comprendre les mécanismes physiopathologiques et de développer des stratégies thérapeutiques efficaces pour FRDA. Les principaux modèles utilisés dans FRDA sont les souris Frda / MCK, Frda / NSE et YG8R.
6.1. Souris Frda / MCK et Frda / NSE
Une délétion totale de la frataxine est létale chez les souris Knock-Out (KO) pour le gène FXN à cause de son importance durant le développement embryonnaire. Afin de pallier à cette létalité embryonnaire, une stratégie permettant d'inactiver le gène FXN dans un tissu spécifique a été entreprise. Cette approche a amené à des modèles murins de cardiomyopathie (Frda / MCK) et de neurodégénérescence (Frda / NSE). La caractérisation de ces deux modèles a été établie par l’équipe de H. Puccio par inactivation conditionnelle spatio-temporelle (système Cre-Lox) du gène
FXN. L’essentiel des caractéristiques physiopathologiques et biochimiques de FRDA a été
reconstitué par ces deux modèles (Puccio, Simon et al. 2001, Simon, Seznec et al. 2004).
La souris Frda / MCK exprime une Cre recombinase sous le contrôle du promoteur d’une kinase musculaire de créatine (MCK, «Muscle Creatine Kinase»). L’absence de frataxine est spécifique aux cellules squelettiques et aux cardiomyocytes ce qui entraine une mort précoce à l’âge de 10 semaines, due essentiellement à une hypertrophie cardiaque sévère. Le modèle murin Frda / MCK représente le seul modèle qui reproduit la pathophysiologie cardiaque de FRDA.
La souris Frda / NSE présente un phénotype plus sévère à la naissance avec une espérance de vie d’une moyenne de 25 jours. Dans ce modèle la Cre recombinase est sous le contrôle du promoteur de l’énolase neuronale (NSE, «Neuron Specific Enolase») reproduisant ainsi les lésions neurologiques. Cependant, il a été rapporté chez ce modèle des lésions touchant d’autres régions du système nerveux et différents tissus non atteints chez les patients FRDA. De ce fait, le nombre important de tissus lésés dans ce modèle l’empêche de l’utiliser pour des essais thérapeutiques.
6.2. Souris YG8R
Le modèle murin Knock-In (YG8R) est caractérisé par l’équipe du Dr. Pook. Dans cette souris, les gènes murins FXN ont été modifiés de telle sorte à les rendre non fonctionnels et un autre gène FXN
20
humain a été introduit. Ce gène provenant d’un patient FRDA contient des séquences nucléotidiques du promoteur, des séquences codant pour la frataxine et une séquence GAA répétée 230 fois dans le 1er intron, exprimant ainsi autour de 25 % du niveau normal de frataxine (Clark, De Biase et al.
2007).
D'autres modèles en cours de développement utilisent la technique de petits ARN inhibiteurs (siRNA) pour diminuer fortement le niveau de la frataxine. Ces modèles ont été faits de manière à pouvoir modifier à la volonté de l'expérimentateur le niveau de frataxine et donc d'évaluer la réversibilité des dégâts causés par le déficit de frataxine. Toutefois, aucun de ces modèles n’est parfaitement adéquat pour répondre totalement aux attentes de la recherche.
7. Les approches thérapeutiques 7.1. Diagnostic prénatal
Depuis 2001, un diagnostic prénatal a pu être proposé pour les couples qui sont à risque et qui veulent concevoir des enfants. La mutation génétique sera recherchée sur les villosités choriales dès 12 semaines d’aménorrhée ou grâce à une amniocentèse sur les cellules amniotiques dès 16 semaines d’aménorrhée. Le diagnostic biologique repose sur l’analyse de l'ADN afin de mettre en évidence l'expansion du triplet GAA par PCR (Polymerase Chain Reaction) ou bien par Southern Blot
(Romeu 2012).
7.2. Thérapie pharmacologique
Cette thérapie repose sur l’utilisation de petites molécules capables de contourner les conséquences fonctionnelles dues au déficit en frataxine chez les patient FRDA. L’idébénone a été le premier composé pharmaceutique utilisé pour traiter les patients FRDA. Son utilisation a déjà donné des résultats prometteurs tant dans des modèles expérimentaux qu’en clinique, du moins sur la cardiopathie de FRDA. Ce composé a été également testé pour ses effets potentiels sur le système nerveux des patients, il est en phase III de développement clinique aux États-Unis et en Europe
(Seznec, Simon et al. 2004) (Tableau 3). D’autres composés ont connu un développement positif et ils sont actuellement en phase II (pioglizatone). Ils visent principalement les fonctions mitochondriales ou une amélioration des mécanismes antioxydants. Enfin, d’autres méthodes proposent une réduction des réserves de fer mitochondriales (deferiprone, phase II).
21 7.3. Thérapie génique et autres perspectives
Cette voie thérapeutique consiste à transférer une copie du gène FXN dans les cellules des patients FRDA et plus particulièrement dans les cellules cardiaques et les neurones. Cette stratégie thérapeutique a déjà été entreprise par l’équipe du Dr. Tremblay et celle du Dr. Puccio sur des modèles de souris reproduisant les symptômes cardiaques et neurologiques retrouvés chez les patients FRDA (Belbellaa and Puccio 2014, Catherine Gérard 2014). Elle est basée sur l’utilisation d’un virus adéno-associé (AAV, «Adeno-Associated Virus»), connu pour cibler et faire exprimer avec efficacité un gène thérapeutique dans les cellules. Le virus a été modifié de manière à perdre son pouvoir pathogène, tout en conservant sa capacité d’acheminer une copie normale du gène FXN dans les cellules et d’y faire ainsi exprimer la frataxine. Cette méthode a non seulement permis de prévenir le développement de problèmes cardiaques mais aussi un rétablissement des fonctions cardiaques des souris. Par ailleurs, il reste à vérifier si cette approche présente la même efficacité au niveau du système nerveux.
Une autre possibilité thérapeutique utilise des molécules qui peuvent augmenter l’expression de la frataxine chez les patients FRDA à des niveaux comparables à ceux retrouvés chez les porteurs. Parmi ces molécules, il y a des protéines TALE (Transcription Activator-Like Effector) ciblant le promoteur de la frataxine (Chapdelaine, Coulombe et al. 2013), les inhibiteurs des histones déacétylase (HDAC) et les polyamides (Chou, Herman et al. 2008, Rai, Soragni et al. 2010).
22
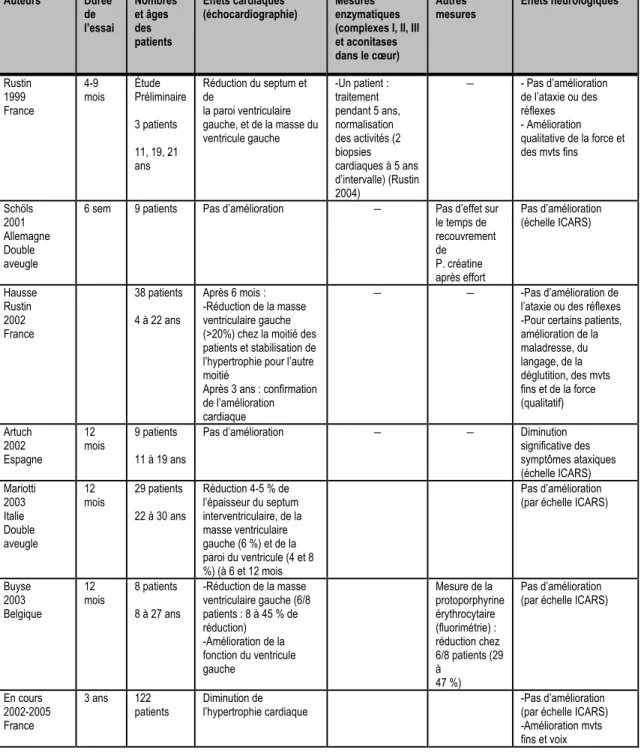
Tableau 2 : Les essais cliniques réalisés avec l’idébénone chez les patients FRDA Échelle ICARS : International Cooperative Ataxia Rating Scales, P. créatine : phosphocréatine, mvts : mouvements.(Tableau tiré de (Simon 2004))
Auteurs Durée de l’essai Nombres et âges des patients Effets cardiaques
(échocardiographie) Mesures enzymatiques (complexes I, II, III et aconitases dans le cœur)
Autres
mesures Effets neurologiques
Rustin 1999 France
4-9
mois Étude Préliminaire 3 patients 11, 19, 21 ans Réduction du septum et de la paroi ventriculaire gauche, et de la masse du ventricule gauche -Un patient : traitement pendant 5 ans, normalisation des activités (2 biopsies cardiaques à 5 ans d’intervalle) (Rustin 2004) − - Pas d’amélioration de l’ataxie ou des réflexes - Amélioration qualitative de la force et des mvts fins Schöls 2001 Allemagne Double aveugle
6 sem 9 patients Pas d’amélioration − Pas d’effet sur le temps de recouvrement de P. créatine après effort Pas d’amélioration (échelle ICARS) Hausse Rustin 2002 France 38 patients 4 à 22 ans Après 6 mois : -Réduction de la masse ventriculaire gauche (>20%) chez la moitié des patients et stabilisation de l’hypertrophie pour l’autre moitié
Après 3 ans : confirmation de l’amélioration cardiaque
− − -Pas d’amélioration de
l’ataxie ou des réflexes -Pour certains patients, amélioration de la maladresse, du langage, de la déglutition, des mvts fins et de la force (qualitatif) Artuch 2002 Espagne 12 mois 9 patients 11 à 19 ans
Pas d’amélioration − − Diminution
significative des symptômes ataxiques (échelle ICARS) Mariotti 2003 Italie Double aveugle 12 mois 29 patients 22 à 30 ans Réduction 4-5 % de l’épaisseur du septum interventriculaire, de la masse ventriculaire gauche (6 %) et de la paroi du ventricule (4 et 8 %) (à 6 et 12 mois Pas d’amélioration (par échelle ICARS)
Buyse 2003 Belgique 12 mois 8 patients 8 à 27 ans -Réduction de la masse ventriculaire gauche (6/8 patients : 8 à 45 % de réduction) -Amélioration de la fonction du ventricule gauche Mesure de la protoporphyrine érythrocytaire (fluorimétrie) : réduction chez 6/8 patients (29 à 47 %) Pas d’amélioration (par échelle ICARS)
En cours 2002-2005 France
3 ans 122
patients Diminution de l’hypertrophie cardiaque -Pas d’amélioration (par échelle ICARS) -Amélioration mvts fins et voix
23 1. Généralité
La recherche dans le développement de peptides ou de protéines à visée thérapeutique est longtemps restée limitée en raison de leur faible perméabilité membranaire et l’acheminement sélectif de molécules à l’intérieur de la cellule. Ces contraintes ont été contournées grâce à la découverte de peptides de pénétration cellulaire (CPP, «Cell Penetrating Peptide») appelés aussi domaines de transduction protéique (PTD, «Protein Transduction Domain»). Ils sont à caractères basiques amphiphiles capables de franchir les membranes cellulaires et d’entrer dans les cellules, seuls ou en transportant des molécules (cargos) auxquels ils sont fusionnés comme les
oligonucléotides, les liposomes, les protéines et bien d’autres (Schwarze and Dowdy 2000, Wadia
and Dowdy 2002). Les CPPs sont de courtes séquences peptidiques qui varient de 7 à 30 acides aminés, naturels ou synthétiques et rentrent à l’intérieur des cellules selon une ou plusieurs voies d’internalisation dépendant et / ou indépendant de l’énergie (Thoren, Persson et al. 2000). Cependant, les CPPs présentent des inconvénients par leur faible spécificité si un ciblage est souhaité car, ils peuvent s’internaliser dans plusieurs types cellulaires bien que leur capacité de pénétration soit variée d’un type cellulaire à un autre (Mai, Shen et al. 2002, Niesner, Halin et al. 2002) (Tableau 4).
Historiquement, les premières observations ont été réalisées sur la protéine Tat dérivée du virus de l’immunodéficience 1 (HIV-1). Il a été démontré que cette protéine purifiée pouvait pénétrer dans les
cellules et se concentrer autour du noyau (Frankel and Pabo 1988, Green and Loewenstein 1988).
Par la suite, les recherches menées par Joliot et ses collègues ont rapporté que l’homéodomaine d’Antennapedia (43-58), une homéoprotéine de drosophile, avait la capacité de pénétrer dans les neurones différentiés tout en stimulant leur croissance (Joliot, Pernelle et al. 1991). De ces travaux émergent la découverte du premier CPP, un peptide de 16 acides aminés correspondant à la troisième hélice de Drosophila antennapedia homeodomain, nommé Pénétratine (Pen) (Derossi, Joliot et al. 1994). Ainsi, la capacité des peptides Pénétratine et Tat de se distribuer au sein des cellules a mené au développement d’une longue série de CPPs comprenant les peptides chimères (CADY (Konate, Crombez et al. 2010), MPG (Crombez, Charnet et al. 2007), Pep-1 (Morris, Depollier et al. 2001) et transportan (Pooga, Hallbrink et al. 1998)), les peptides modèles (polyarginine (Zhang,
24
Tang et al. 2006)) et les peptides naturels (maurocalcine et crotamine (Radis-Baptista, de la Torre et al. 2008)). Le peptide MPG était le premier CPP permettant la délivrance d’acides nucléiques (Morris, Vidal et al. 1997). Par ailleurs, cette même stratégie a été adoptée pour la délivrance des protéines et des peptides par Pep-1 (Morris, Depollier et al. 2001). Pour l’application des CPPs in vivo, le groupe de Dowdy a rapporté que Tat pouvait délivrer la β-galactosidase dans plusieurs organes chez la souris et traverser la barrière hémato-encéphalique (BHE) (Schwarze, Ho et al. 1999).
Les CPPs sont de plus en plus utilisés comme outils pour l’internalisation in vitro et in vivo de nombreuses molécules biologiquement actives. De plus, la conception de plusieurs analogues est possible du fait de leur simple accessibilité par voie de synthèse (Schwarze, Ho et al. 1999, Lindgren, Hallbrink et al. 2000).
25 Tableau 3 : Les propriétés de quelques peptides de pénétration cellulaire (CPPs)
(Tableau tiré de (Poillot and De Waard 2011))
Nom Origine Séquence peptidique Référence
CPP naturels Pénétratine
(pAntp) Protéine Antennapedia de la Drosophile
RQIKIWFQNRRMKWKK (Derossi et al., 1994)
Tat (48-60) Protéine Tat du
VIH GRKKRRQRRRPPQ (Vives et al., 1997)
MCa Toxine du «Scorpio
maurus palmatus»
GDCLPHLKLCKENKDCCSKKCKRR
GTNIEKRCR (Estève et al., 2005)
VP22 Protéine de
l’enveloppe du Virus Herpès Simplex
DAATATRGRSAARPTERPRAPARS
ASRPRRPVE (Elliott et O'Hare 1997)
hCT(9-32) C alcitonine
humaine LGTYTQDFNKFHTFPQTAIGVGAP -amide (Schmidt et al., 1998)
PrP Protéine du prion
de souris MANLGYWLLALFVTMWTDVGLC KKRPKP (Lundberg et al., 2002) CPP synthétiques
Transportan
Galanin(1-12)-LysMastoparan GWTLNSAGYLLGKINLKALAALA KKIL-amide (Pooga et al., 1998)
TP10 Transportan
tronqué AGYLLGKINLKALAALAKKIL-amide (Soomets et al., 2000)
MAP Peptide
Amphipathique modèle
KLALKLALKALKAALKLA-amide (Oehlke et al., 1998)
Pep-1 Séquence NLS +
Séquence hydrophobe
KETWWETWWTEWSQPKKKRKV-cys amide (Morris et al., 2001)
MPG Séquence NLS +
Séquence hydrophobe
GALFLGWLGAAGSTMGAPKKKR
KV-cys amide (Morris et al., 1997)
Oligoarginine (R)n (Rothbard et al.,
26
2. Mécanismes d’entrée dans la cellule
Les mécanismes moléculaires fondamentaux qui conduisent à l’internalisation des CPPs ne sont pas encore clairement identifiés, malgré leur potentiel pour le développement et l’optimisation de stratégies à visée thérapeutiques. Il est admis que la première interaction entre les CPPs et les cellules implique des liaisons électrostatiques avec les protéoglycanes de la surface cellulaire, suivie d’une réorganisation des phospholipides pour permettre l’entrée des CPPs par voie d’endocytose ou par translocation directe à travers la membrane plasmique (Heitz, Morris et al. 2009) (Figure 7).
2.1. La translocation directe
C’est un processus énergie-indépendant (Pooga, Lindgren et al. 1998), dans lequel les CPPs empruntent un passage direct et rapide à travers la membrane cellulaire via la formation de pores qui perturbent l’organisation de la bicouche lipidique. Elle peut suivre deux modèles, le modèle de Barrel-Stave et le modèle de Carpet (Yeaman and Yount 2003, Herbig, Weller et al. 2005). La formation des pores de Barrel-Stave implique d’abord l’insertion des CPPs à la surface membranaire dans une orientation perpendiculaire de façon à positionner leurs résidus hydrophobes face à la bicouche lipidique. Par la suite, les CPPs se regroupent jusqu’ à atteindre un certain niveau de concentration formant ainsi un canal ayant l’aspect d’un tonneau pour se retrouver enfin dans le cytoplasme (Yeaman and Yount 2003). Dans le modèle de Carpet, la membrane plasmique est tapissée par toute la longueur des CPPs de manière à établir des liaisons électrostatiques avec les têtes des phospholipides sans formation de pores à la membrane. Lors de ce mécanisme, il se produit une réorganisation et une courbure de la bicouche lipidique menant à l’entrée des CPPs dans le
cytoplasme (Yeaman and Yount 2003, Sanderson 2005).
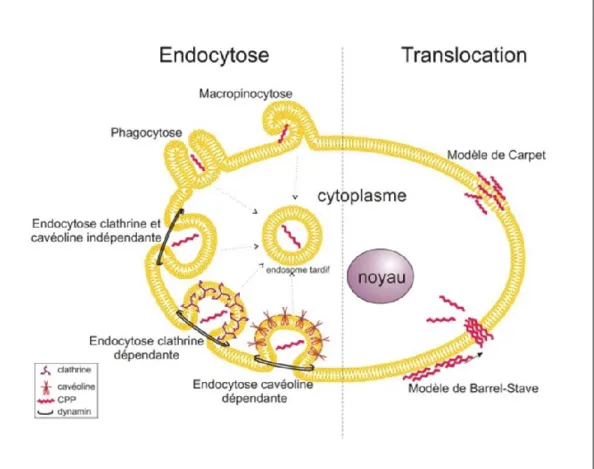
2.2. L’endocytose
C’est un processus d’ingestion cellulaire énergie-dépendant utilisé pour internaliser une large variété de macromolécules à travers la membrane (Watkins, Schmaljohann et al. 2009). Elle regroupe la phagocytose pour les particules volumineuses et la pinocytose pour les fluides et les solutés. La phagocytose est un mécanisme spécifique aux cellules spécialisées comme les macrophages, les cellules dendritiques et les neutrophiles. La pinocytose concerne différents types cellulaires et peut être subdivisée en quatre groupes: la macropinocytose, l’endocytose dépendante de la cavéoline ou
27
de la clathrine et l’endocytose indépendante de la clathrine et de la cavéoline (Conner and Schmid 2003) (Figure 6).
La contribution apportée par les deux mécanismes d’internalisation des CPPs (translocation versus endocytose) peut varier en fonction de la nature du CPP, du cargo auquel il est fusionné, du type cellulaire et des concentrations expérimentales utilisées. Il a été aussi accepté que la plus part des CPPs ne sont pas internalisés par une seule voie mais que l’endocytose et la translocation directe peuvent intervenir simultanément et à des proportions différentes (Jiao, Delaroche et al. 2009).
28
Figure 6 : Les mécanismes d’entrée dans la cellule des peptides de pénétration cellulaire (Image tirée de (Poillot and De Waard 2011))
29 3. Exemples de CPP
3.1. Le peptide Tat
La protéine Tat (Trans-Activateur de transcription) est codée par le virus de l'immunodéficience humaine (VIH-1). Elle est nécessaire pour la réplication et la régulation en se liant sur l’élément TAR (Trans-Activator Responsive element), une séquence présente à l’extrémité 5’ de l’ARN messager viral (Brigati, Giacca et al. 2003). En plus de ses fonctions transcriptionnelles, Tat semble avoir la capacité de se libérer des cellules infectées par le VIH-1 et d’interagir avec les cellules voisines non infectées en favorisant l’internalisation et la duplication du virus tout en gardant sa propriété trans-activatrice (Frankel and Pabo 1988). De plus, elle induit la production de la cytokine IL-10 qui est un élément important dans l’évolution de la maladie vers le stade SIDA (Syndrome de l’Immunodéficience Acquise). La protéine Tat manifeste également un pouvoir pathogène en modulant l’apoptose des cellules et en stimulant l’expression des co-récepteurs du VIH (Xiao, Neuveut et al. 2000, Vasilescu, Heath et al. 2003) .
En 1994, le couplage d’une séquence peptidique de Tat (37-72) à la β-galactosidase et l’injection de ce complexe à des souris par voie intraveineuse permettait la délivrance du complexe dans le foie, la rate et le cœur, et de façon moindre, dans les muscles squelettiques, les poumons et les reins
(Fawell, Seery et al. 1994). Cette étude a révélé la séquence peptidique minimale de Tat qui lui confère la capacité de pénétration cellulaire. Elle contient les acides aminés (47-60) appartenant au cluster basique de la protéine (Vives, Brodin et al. 1997). Afin de déterminer la voie d’entrée de Tat, les premières études étaient basées sur l’utilisation de microscopie de fluorescence sur des cellules fixées et de cytométrie en flux, suggérant que le mécanisme d’internalisation cellulaire du peptide Tat suivait une voie indépendante de l’énergie, de l’endocytose et de transporteurs ou de récepteurs spécifiques à la surface des cellules.
Ainsi, en 2003 Richard et ses collègues ont réévalué le mécanisme d’internalisation de Tat. Ils ont démontré que le peptide emprunte réellement une voie dépendante de l’endocytose en se localisant principalement au niveau des endosomes et que les résultats obtenus initialement sont dues aux artéfacts associés aux techniques expérimentales (Richard, Melikov et al. 2003). L’étape critique pour le peptide et son cargo après l’internalisation est la sortie endosomale afin de parvenir à sa cible intracellulaire. Cette étape sollicite l’utilisation d’agents pour favoriser l’acidification de
30
l’endosome sans quoi, le cargo reste à l’intérieur de l’endosome et est dégradé par fusion avec les lysosomes. L’utilisation de Tat comme vecteur d’internalisation se distingue par sa remarquable capacité d’être attaché à des cargos d’une structure allant jusqu’à 200 nm de diamètre (liposome). Le couplage à des protéines peut être réalisé sur chacune des extrémités, côté N-terminal ou C-terminal par l’intermédiaire d’un pont disulfure dans la plupart des cas. À ce jour, plusieurs molécules couplées au peptide Tat ont été transportées à l’intérieur des cellules, comme des agents de contraste ou des sondes fluorescentes pour l’imagerie médicale, des siARN ou des agents chimiothérapeutiques.
3.2. Le peptide Pep-1
C’est un peptide vecteur amphipathique primaire de nature synthétique. Il est composé de 21 acides aminés et comporte trois domaines distincts : un domaine hydrophobe riche en tryptophane (Trp) du côté N-terminal qui permet des interactions stables non covalentes avec le cargo et la membrane cellulaire (Crombez, Morris et al. 2009), un domaine hydrophile, dérivé de la séquence NLS du virus à ADN SV40, riche en résidus lysine (Lys) du côté C-terminal qui facilite l’internalisation cellulaire et un court bras espaceur constitué d’une sérine (Ser), d’une glutamine (Gln) et d’une proline (Pro) assurant une certaine flexibilité entre le cargo et le peptide pour la délivrance du cargo (Deshayes, Morris et al. 2008).
Pep-1 pénètre dans les cellules efficacement pour se localiser majoritairement autour du noyau suivant un mécanisme d’internalisation non endocytaire (Munoz-Morris, Heitz et al. 2007). Ce peptide piège la molécule d’intérêt dans un réseau peptidique formant un complexe stable et limite la dégradation de la molécule à transporter. Commercialisé sous le nom de Chariot, Pep-1 a été appliqué pour la vectorisation de peptides dérivés de Cdc25C ou de HIV-1 mais également d’inhibiteurs protéiques pour le contrôle de la progression du cycle cellulaire (Morris, Depollier et al. 2001, Munoz-Morris, Heitz et al. 2007). Actuellement, Pep-1 est utilisé principalement pour la livraison de peptides, protéines, PNA et anticorps dans plusieurs lignées cellulaires humaines ou modèles animaux par différentes méthodes d’administration.