OATAO is an open access repository that collects the work of Toulouse
researchers and makes it freely available over the web where possible
Any correspondence concerning this service should be sent
to the repository administrator:
[email protected]
This is an author’s version published in:
http://oatao.univ-toulouse.fr/27596
To cite this version:
Mayen, Laëtitia
and Jensen, Nicholai D. and Laurencin, Danielle and
Marsan, Olivier
and Bonhomme, Christian and Gervais, Christel and Smith,
Mark E. and Coelho, Cristina and Laurent, Guillaume and Trebosc, Julien and
Gan, Zhehong and Chen, Kuizhi and Rey, Christian
and Combes,
Christèle
and Soulié, Jérémy
A soft-chemistry approach to the synthesis of
amorphous calcium ortho/pyrophosphate biomaterials of tunable composition.
(2020) Acta Biomaterialia, 103. 333-345. ISSN 1742-7061
A soft-chemistry approach to the synthesis of amorphous calcium
ortho/pyrophosphate biomaterials of tunable composition
Laëtitia Mayen
a
.
1, Nicholai D. Jensen
b
,
c,
1
,
Danielle Laurencin
b
, Olivier Marsan
a,
Christian Bonhomme
c, Christel Gervais
c, Mark E. Smith
<l, Cristina Coe
l
ho
c,
Guillaume Laurent
',
Julien Trebosc
e, Zhehong Gan
r
. Kuizhi Chen
r
. Christian Rey
a
,
Christèle Combes
a, Jérémy Soulié
a,
+•C/RJMAT. Université de Toulouse, CNRS, INPT-ENSIACET, Toulouse, France b /CGM, CNRS-UM-ENSCM, Université de Montpellier; Montpellier; France <Sorbonne Université, CNRS, LCMCP. Paris, France
d Depamnenr of Chemisrry, Lancaster Universiry, Lancaster, UK
• Université de Lille, UMR 8181, UCCS: Unir of Caralysis and Chemisrry of So!ids, Lille, France
r National High Magneric Field Laborarory, Tallahassee, FL, USA
ARTICLE INFO
Keywords:
Amorphous materials
Mixed calcium orrho/pyrophosphate Soft chemistry
Biomaterials
• Corresponding author.
ABSTRACT
The development of amorphous phosphate-based materials is of major interest in the field of bioma terials science, and especially for bone substitution applications. ln this context, we herein report the synthesis of gel-derived hydrated amorphous calcium/sodium ortho/pyrophosphate materials at ambi ent temperature and in water. For the first time, such materials have been obtained in a large range of tunable orthophosphate/pyrophosphate molar ratios. Multi-scale characterization was carried out thanks to various techniques, including advanced multinuclear solid state NMR. lt allowed the quantifi cation of each ionic/molecular species leading to a general formula for these materials: [(Ca2+y Na+, H+3+"-2y-z)(P043-)
1.x(P2O74-),d(H2O)u. Beyond this formula, the analyses suggest that these amorphous
solids are formed by the aggregation of colloids and that surface water and sodium could play a rote in the cohesion of the whole material. Although the full comprehension of mechanisms of formation and structure is still to be investigated in detail, the straightforward synthesis of these new amorphous ma terials opens up many perspectives in the field of materials for bone substitution and regeneration.
Statement of significance
The metastability of amorphous phosphate-based materials with various chain length often improves their (bio )chemic.al reactivity. However, the contrai of the ratio of the different phosphate entities has not been yet described especially for small ions (pyrophosphate/orthophosphate) and using soft chem istry, whereas it opens the way for the tuning of enzyme- and/or pH-driven degradation and biologi cal properties. Our study focuses on elaboration of amorphous gel-derived hydrated calcium/sodium or tho/pyrophosphate solids at 70 °C with a large range of orthophosphate/pyrophosphate ratios. Multi-scale characterization was carried out using various techniques such as advanced multinuclear SSNMR
(3
1 P,23Na, 1 H, 43Ca). Analyses suggest that these solids are formed by colloids aggregation and that the loca tion of mobile water and sodium could play a rote in the material cohesion.
1. Introduction
E-mail addresses: [email protected], [email protected] U Soulié).
Among inorganic materials for bone substitution, amorphous
solids have attracted a lot of attention. Indeed, their metastability
often improves their (bio)chemical reactivity. The subsequent re
Iease of active ions
(1 ]
, and/or dissolution/precipitation reactions
Iead to neoformed apatite, that positively impacts
osteoconduc-1 These authors contributed equally to the work.
tion andosteoinduction [2] . The most famous example of amor-phousbiomaterialsisprobablythatofthebioactivesilicate-based glasses (either sol-gel or melt-derived), which have been exten-sively studied [2 ,3] . The presentpaper, however, focuseson new typesofphosphate-basedamorphousmaterialsforbone regenera-tion.Twomainfamiliesofmaterialsbelongingtothiscategoryare studiedby different scientific communities andare referenced in theliteratureasphosphate-basedglassesandamorphouscalcium phosphates.
On theone hand,phosphate-basedglassesare generallymade byfusion. Consideringpure binary P2O5–CaOglasses [4] , the
na-tureofthephosphateunits(asexpressed bythe Qn terminology,
wheren is the numberof bridging oxygen atoms per PO4
tetra-hedron)canbe controlledthrough theatomicCa/P ratio.This ra-tiocanvaryfromultraphosphates(phosphate3D networkmainly basedonQ3 species) tometaphosphateglasses(linear phosphate
chains, mainly Q2) and eventually to invert glasses(isolated
py-rophosphatesandorthophosphates, Q1 andQ0 respectively). This
rangeofcomposition andthe useofadditives allowthe elabora-tionofsuchglassesandthecontroloftheirdissolution[5–9] .
Ontheotherhand,amorphouscalciumphosphatepowderscan be synthesizedby precipitation in solutionat room temperature,
most of the time in water and without further high
tempera-turetreatment [10] . These are generallyobtained by double de-composition betweensolublecalcium andphosphate salt precur-sors,and some processes lead to dense liquid“coacervates” [11] . Severalkindsofphosphateprecursorshavebeenused: long-chain polyphosphates(polyP)[12 ,13] ,cyclicpolyphosphates[14] , diphos-phates(alsocalledpyrophosphates:P2O74−)[15–17] or
orthophos-phates(PO43−) [10] .Withoutanyhightemperaturetreatment,the
polyPassociationsare preservedinthe final materials, which re-mainamorphous. Incontrast,crystallizationcanoccur whenonly orthophosphates orpyrophosphates (smaller anions) are present. Inthiscase,theamorphous/crystallinenaturedependsonthe syn-theticparametersused(pH,concentration…)[16–18] .
In thiscontext, werecentlyreportedthesynthesis ofdifferent compositionsofmonolithiccalciumandpotassiumpyrophosphate materials (PYG materials, PYrophosphate Glasses) preparedusing softconditions(in aqueoussolution witha dryingstep at70°C) [17] .Weshowedthat anincrease oftheCa2+/P2O74− ratiointhe
precursorbatchsolutionresultedinanincrease oftheproportion ofcrystalline calcium pyrophosphate phase in the final material. Thisbehavior was suggestedto be induced by small amounts of orthophosphateionsformedbypartialhydrolysisoftheinitial py-rophosphateentitiesduringthesynthesisprocess,whichinhibited calciumpyrophosphatecrystallization.
Considering thisresult, the aimofthis paperisto presentan originalstrategy forthe low-temperaturesynthesis ofgel-derived amorphouscalciumphosphatematerials (NaPYG)containingboth pyrophosphateandorthophosphateentitiesincontrolledamounts,
and calcium and sodium as metal cations. To the best of our
knowledge, this approach has never been described previously. Moreover,sodiumphosphatesaltswerechosenhere(inpreference tothepotassiumones),becausesodiumexhibitsnosideeffectsin vivolikehyperkalemia[19] (contrarytopotassium).
Beyond the novelty of this synthesis, these mixed
or-tho/pyrophosphate materials appeared to us as potentially at-tractivecandidatesforbone substitution applications.Indeed,the interest of pure calcium pyrophosphate biomaterials has been demonstrated in the literature, through acellular and cellular in vitrotests[20 ,21] ,in vivo animalstudies [22] ,anda clinical trial [23] . Pyrophosphates have been shown to hydrolyze in vivo by enzymatic reactions [20] and/or acidic pH [24] (due to inflam-matory response after implantation), leading to orthophosphate ions, which are one of the key “building ions” of bone mineral. Pyrophosphate is one of the mineral ions considered to inhibit
mineralformation.Inosteoblastculturesespecially,pyrophosphate inhibition occurs by binding to the mineral,up-regulating osteo-pontin, and inhibiting alkaline phosphatase activity [25] . It has been observed,however, that, in vivo,the pyrophosphatelevel is ratherwell controlled andthat its variations, dueto physical ac-tivityforexample,are regulated[26] .More generally,inhumans, the total production ofpyrophosphate ions per day is evaluated, from0.7toseveralkg[26] andmorethat170 biologicalreactions involvingpyrophosphateionshavebeenidentified[26] .
Therefore,the controlofthe ortho/pyrophosphateratiowithin anamorphousmaterial(synthesizedatatemperatureofonly70°C andwithoutanyadditives)couldpotentially beaninteresting ad-justable parameterfortuning thekineticsofbiomaterial degrada-tionandbonemineralformation.
In this work, we thus investigate in detail the effect of the ortho/pyrophosphate content on the nature, structure and mor-phologyofnewly-synthesizedamorphousbiomaterials,using com-plementarycharacterizationtechniquesandinparticularadvanced multinuclear solid state NMR. Advanced solid state NMR are of paramount importance in the studies of amorphous derivatives suchasbioglassesforwhichXRDdataaremuchlessinformative. The smart use of the NMR interactions (indirect Jcouplings and spatial dipolar couplings, D) allows to safely establish through-bond andthrough-space connectivities. In thiswork, 31P INADE-QUATEMAS experimentswere exploredtodisentanglethe contri-butionsofortho- andpyrophosphatesgroupsin1D 31P MAS ex-periments(especiallyforamorphousmaterials).Mostimportantly,
homo- (31P-31P) and heteronuclear (23Na-31P) 2D correlation
experimentswereimplementedto highlightthepresence/absence
of segregated domains (both from the anionic, ie ortho- and
pyrophosphates,andcationic,ieNa+andCa2+,pointsofview).
2. Experimentalsection
2.1. Precursors
Calciumchloridedihydrate(CaCl2•2H2O,Merck)andtri-sodium
phosphate dodecahydrate (Na3PO4•12H2O, GRP rectapur, VWR
Chemicals) were used as received, as calcium and
orthophos-phate sources, respectively. The pyrophosphate precursor, anhy-drous tetrasodium pyrophosphate (Na4P2O7), was prepared by
heating disodium hydrogen phosphate powder (Na2HPO4, VWR
Chemicals)at400 °Cduring15h inamufflefurnace.The forma-tion of thissalt wasverified by XRD,Raman and 31Psolid state
NMR spectroscopies before its use (in particular, no residual or-thophosphateentitywasdetectedbyNMR).
2.2. Synthesis
Calciumandphosphatereagentsolutionswere prepared sepa-ratelybydissolvingCaCl2•2H2Oin40mLofdeionizedwater
(solu-tion A), andorthophosphate (Na3PO4•12H2O) andpyrophosphate
(Na4P2O7) precursors in400 mL ofdeionized water (Solution B).
SolutionBwaspreparedwithdifferentmolarratiosof orthophos-phateandpyrophosphateions(PO43−/(P2O74− +PO43−)),inorder
tostudytheinfluenceofthisparameteronthenatureand compo-sitionofthefinalmaterial.SamplesarelabeledNaPYG-0× 0,from the lowest (X = 0) to the highest (X = 6) ortho/pyrophosphate ratio (Table 1 ). Solution Awas then added tosolution B using a peristalticpumpata constantvolumetricflowrate(32 Mlmin−1) forallsamples.Themixtureturnedtransparenttotranslucent. Af-tercompleteaddition,thesolutionwasstirredfor5moreminutes (aliquotsofthesolutionwerecollectedduringthisstepinorderto measure thepH), andthefinal colloidalsolution wascentrifuged 5minat7500 rpm.Theresultingdense gelatthe bottomofthe
Table 1
Number of moles of calcium and phosphate salt precursors involved in the initial solutions (A and B) and A and B solution volumes. Molar ratio, percentage, and pH after mixing the two solutions are also reported.
NaPYG 0 0 0 NaPYG 010 NaPYG 020 NaPYG 030 NaPYG 040 NaPYG 050 NaPYG 060 CaCl 2 • 2H 2 O (mmol) 7.210 ± 0.020
Na 4 P 2 O 7 (mmol) 33.30 ± 0.008 28.300 ± 0.008 23.310 ± 0.008 16.650 ± 0.008 9.990 ± 0.008 4.990 ± 0.008 0
Na 3 PO 4 • 12H 2 O (mmol) 0 4.990 ± 0.006 9.99 ± 0.005 16.65 ± 0.006 23.31 ± 0.006 28.30 ± 0.006 33.330 ± 0.006 Calcium solution (A)
volume (mL)
40.0 ± 0.1 Phosphate solution (B)
volume (mL) 400.0 ± 0.1
Ca/P (molar ratio) 0.109 ± 0.003 0.119 ± 0.004 0.128 ± 0.003 0.145 ± 0.003 0.166 ± 0.003 0.190 ± 0.004 0.220 ± 0.003 Orthophosphate initial
molar%:
PO 43−/(P 2 O 74−+ PO 43−)
0 15.00 ± 0.25 30.00 ± 0.14 50.00 ± 0.11 70.00 ± 0.12 85.00 ± 0.20 100
pH after mixing A and B solutions
10.1 ± 0.1 11.5 ± 0.1 11.8 ± 0.1 11.9 ± 0.1 12.0 ± 0.1 12.1 ± 0.1 12.1 ± 0.1
centrifugetubewaswashedthreetimeswithdeionizedwater. Fi-nally,thewashedgelwaspouredintoaglasscrystallizingdishand driedat70°Cduring7days.Sampleswerethenstoredat−20°C beforetheircharacterization(carriedoutatambienttemperature).
α
-canaphite (Na2CaP2O7•4H2O) and nanocrystalline apatitewere prepared foruse as referencecompounds forRaman spec-troscopy,XRD,andsolidstateNMRcharacterizations,becausethey were occasionally identified in some of the final materials. The nanocrystalline apatite referencesamplewas synthesized accord-ing to a previously published protocol[27] . The canaphite refer-ence samplewas prepared by adapting the protocol reported by Chengetal.[28] (SI-1).Thesetworeferencesampleswere charac-terizedbysolidstate NMRandRamanspectroscopyandXRD,and weredemonstratedtobepurecompounds.
2.3. Characterization
The synthesized materials were characterized using comple-mentary structural, microstructural, elemental, spectroscopic and thermalcharacterizationmethods.
Powder X-ray diffraction analysis was performed using a diffractometer (Bruker D8 advanced) with a copper anticathode (
λ
(Kα1) = 1.54056 ˚A,λ
(Kα2) = 1.54433 ˚A), stepsize of 0.03°be-tween10° and70° Sampleswerecrushedwithoutsievingina mor-tarbeforemeasurements.
Magic-angle spinning (MAS) solid state NMR experiments were performed to analyze 31P, 23Na,1H and 43Ca local environments
in the materials. 1H single pulse, 23Na single pulse, 31P single
pulse, 1H→31P CP (Cross-Polarization) and 31P CP INADEQUATE
[29] (IncredibleNaturalAbundanceDoublEQUAntumTransfer
Ex-periment) spectra were recorded on a VNMRS-600 MHz (14.1 T)
instrument equipped with a Varian T3 3.2 mm triple resonance probeusing14to20kHzspinningspeeds.1Hsinglepulseand1H
Hahnechospectrawereacquiredat14.1TusingaVarian1.2mm triple resonanceprobe spinningat40 kHz.1H{31P}HETCOR
(HET-eronuclear CORrelation)spectra andadditional1H Hahn-echo
ex-perimentswere alsorecordedat14.1Tusinga1.6mmtriple res-onance probe, with 22 to 30 kHz spinning speeds. 31P CP
SQ-DQ(SingleQuantum-DoubleQuantum)experimentswithSPC5
re-couplingwere performedon a 700MHz (16.4T) Bruker AVANCE
III spectrometer, equipped with a 4 mm double resonance MAS
probe spinningat14kHz[30] .The23Na{31P}D-HMQC
(Heteronu-clearMultiple-QuantumCorrelation)[31] spectrawereacquiredon a800MHzBrukerAdvanceNEO4spectrometer(18.8T)equipped
with a 3.2 mm HXY MAS probe using 20 kHz spinning speed.
43Ca multi-DFS(DoubleFrequencySweep) NMR[32] spectrawere
acquired on a 850 MHz Bruker NEO 4 spectrometer (20.0 T), a
800 MHz Brukeradvance NEO spectrometer (18.8T), orthe 35.2 TSCHmagnetinTallahassee(FL-USA,usingaBrukerAVANCENEO
console),respectivelyequippedwitha7mm× lowgammaprobe,
a 4 mm HX Tallahassee probe and a 3.2 mm MAS Tallahassee
probeusing5to10kHzspinningspeed[33 ,34] .Thecompleteset ofacquisitionparameters canbe foundinsupporting information (SI-2), including information on temperature regulation and the referencingofspectra,anda tablewithall acquisitionparameters (TableS1).
Raman scattering analyses were performed using a Raman LabramHR800confocal microscopeHoribaYvon Jobin.The
sam-ple was exposed to continuous laser radiation provided by a
532 nm Argon diode laser with a power of 14 mW. The
analy-seswerecarriedoutunderaBX41Olympusmicroscopeequipped witha×100lenswithanumericalapertureof0.9, whichconfers to thesystem a lateralresolution of1.0 μm andan axial resolu-tionof 4.5μm.The spectrumof each microdomainwasacquired througha gratingof600lines permm withaspectral resolution of1.5cm−1andcollectedwithaquantumwelldetectorcooledat −60°C by doublePeltiereffect(CCD Synapse). Acertified silicon standardwasusedtocalibratethefrequencyoftheequipment us-ingthe firstorder siliconlineat520.7 cm−1.Each spectrumwas acquiredwithanintegrationtimeof30sand5accumulations.The methodology for Raman lines decomposition to evaluate the or-tho/pyrophosphateratio inthesynthesizedmaterials isdescribed intheSupportingInformation(SI-3,TableS2).
ScanningElectronMicroscopy(SEM)analyseswereperformedon a LEO 435VPmicroscope with an acceleratingvoltage inthe 8– 12 kVrange.Except for NaPYG-000, all sampleswere crushed in amortaruntilgettingsubmillimetricgrains(withoutsieving)that were stuck on adhesive carbon discs and finally silver sputter-coatedbeforeobservation.
ThermogravimetricAnalyses(TGA)andDifferentialThermal Analy-sis(DTA)wereperformedusingaSetaraminstrument(Setsys Evo-lutionSystem)from25to600°Cwithastageof20minat600°C, andaheatingrateof4°Cperminuteinairflow.
InductivelyCoupledPlasma-OpticalEmissionSpectrometryICP-OES
(UltimaExpertmachine)wasusedtoanalyzesolutionsofdissolved powdersin orderto determine theCa, Pand Nacontents in the materials(
λ
Ca=318.12nm,λ
P=177.43nm,λ
Na=589.59nm).UltraHigh Pressure Phase Liquid Chromatography (UHPLC) cou-pledwithanEvaporativeLightScatteringDetector(ELSD)wasused forthedetectionofchlorideions(usinga WatersAcquity appara-tus). The analyzed solutions were the same asfor ICP-OES spec-trometry analysis.Standardcalibration solutions were used (with [Cl−]from0to1.5ppm).
2.4.Statisticsanderrors
Uncertainties in Table 1 are due to volume, mass and pH measurement errors and subsequent propagation of uncertainty.
Table 2
Percentage of phosphorus atom involved in orthophosphate and pyrophosphate ions introduced as precursors in the initial solutions and in the final materials measured by 31 P solid state NMR and Raman spectroscopy.
Precursors in the initial solution 31 P solid state NMR of materials Raman spectroscopy of materials % P pyro % P ortho % P pyro % P ortho % P pyro % P ortho
NaPYG-000 100 0 100 ± 0 0 ± 0 100 ± 0 0 ± 0 NaPYG-010 92 8 74 ± 3 26 ± 3 67 ± 2 33 ± 2 NaPYG-020 82 18 58 ± 3 42 ± 3 52 ± 1 48 ± 1 NaPYG-030 67 33 50 ± 4 50 ± 4 47 ± 1 53 ± 1 NaPYG-040 46 54 37 ± 3 63 ± 3 37 ± 2 63 ± 2 NaPYG-050 26 74 26 ± 6 74 ± 6 31 ± 4 69 ± 4 NaPYG-060 0 100 0 ± 0 100 ± 0 0 ± 0 100 ± 0 Table 3
Chemical composition (Ca, P and Na) of the synthesized NaPYG materials deter- mined by ICP-OES (moles/100 g of dissolved material) and total weight percent of water determined by TGA.
Samples Ca (mol/100 g) P (mol/100 g) Na (mol/100 g) H 2 O w% NaPYG-000 0.320 ± 0.003 0.591 ± 0.004 0.583 ± 0.003 20.1 ± 0.5 NaPYG-010 0.620 ± 0.003 0.599 ± 0.004 0.100 ± 0.001 13.7 ± 0.5 NaPYG-020 0.645 ± 0.004 0.581 ± 0.004 0.100 ± 0.001 12.7 ± 0.5 NaPYG-030 0.669 ± 0.003 0.568 ± 0.005 0.060 ± 0.001 15.3 ± 0.5 NaPYG-040 0.686 ± 0.003 0.545 ± 0.002 0.030 ± 0.001 15.6 ± 0.5 NaPYG-050 0.672 ± 0.006 0.520 ± 0.002 0.030 ± 0.001 15.0 ± 0.5 NaPYG-060 0.772 ± 0.005 0.540 ± 0.005 0.020 ± 0.001 4.9 ± 0.5
Uncertainties in pyrophosphate, orthophosphate (Table 2 and Fig. 7 ) calcium, phosphorus and sodium (Table 3 ) quantification correspond to the standard deviation (triplicate) determined on
values obtained by Raman and NMR spectra deconvolution and
ICPmeasurements. Uncertaintyassociated towaterquantification (Table 3 ) is the nominal value associated to the TGA apparatus. Thoseassociated tothe final composition,charges andthe corre-spondingsummations(Fig. 5 ) havebeencalculatedbythe propa-gationofuncertaintywithpreviousresults.
3. Results 3.1.Morphology
Seven calciumphosphatematerials(NaPYG-000toNaPYG-060)
were prepared in water under mild conditions, using a fixed
amountofCa2+ anddifferentmolarratiosoforthoand
pyrophos-phate entities: PO43−/(P2O74− + PO43−) (Table 1 ). Although the
synthesized materials were obtained as powders for all synthe-sisconditions,theyappearedvisuallydifferent:afinepowderwas observedforNaPYG-000 (preparedfromonlypyrophosphate pre-cursor solution), whereas grains of several millimeterssize with mechanical cohesion were observed for the other compositions (prepared from mixed ortho and pyrophosphate precursor solu-tions). Scanning Electron Microscopy observations (Fig. 1 ) show that:NaPYG-000iscomposedofgrainswithdiametersbetween20 and100μm.Thesegrainsareeitherspherulitesorbundlesformed byplate-likecrystalsofaround2μmwidthand0.5μmthickness. Fortheothersamples,aftergrinding,theparticlesappeartohave angularshapes, with smooth andcompact fracture surfaces with irregular morphologies. These conchoidal fractures are typical of brittlematerials[35] .Fig. 1 (j,k,l)showsanincreaseinthe submi-cronicsurfaceroughness,associatedwithanincreaseoftheinitial ortho/pyrophosphate molarratio in solution (from NaPYG-010 to NaPYG-060).
3.2.Structuralanalyses
The nature of the phases involved was determined by X-ray diffraction analysis (Fig. 2 -a). Samples NaPYG-010 to 050,
pre-paredfromsolutions containingbothpyro- andorthophosphates, were found to be amorphous(very broad halobetween 26° and 34°). On the contrary, well-defined crystalline peaks were ob-served for NaPYG-000. These peaks were identified as those of
α
-canaphite[36] ,Na2CaP2O7•4H2O.X-raydiffractogramofNaPYG-060, showed features of a nanocrystalline apatite [18] , although the apparent crystallite sizes (length and thickness) should be different than that of the nanocrystalline apatite reference sam-ple (3 days of maturation, non-carbonated). Indeed, L(200) (giv-ing informationonthe length ofapatite nanocrystals)andL(310) (givinginformationonthethicknessofapatitenanocrystals)have been calculated using Scherrer’s law. These values are respec-tively: L(200)ref = 17.2 ± 0.6 nm, L(310)ref = 8.3 ± 0.6 nm,
L(200)NaPYG060=29.2± 0.6nm,L(310)NaPYG060=9.2± 0.6nm.
For NaPYG materials, six main domains in the corresponding Raman spectra (Fig. 2 -b) could be distinguished for the phos-phate/pyrophosphateentities[16] :
(i) One domain between 410–650 cm−1 corresponding to vi-brationalbandsofbothorthophosphateandpyrophosphate ions(
δ
PO3andρ
PO3 ofP2O7 andν
4PO4).(ii) Four domains assigned to the pyrophosphateentities only,
including the bending mode of the POP bridge (
δ
POP)around350cm−1;the symmetricstretchingofPOP(
ν
sPOP,between695and795cm−1)andtheintenselinesof
ν
sPO 3,between1015and1070cm−1andof
ν
asPO3 between1080
and1195cm−1.
(iii) Onedomaincorrespondingmainlytothesymmetric stretch-ing of orthophosphate entities (
ν
sPO4) between 915 and
990cm−1.
ForNaPYG-000andNaPYG-060,onlythebandsassociatedwith
pyrophosphate or orthophosphate ions were observed,
respec-tively, as expected. Moreover, these bands, especially
ν
sPO 3(or-thophosphate)and
ν
sPO4 (pyrophosphate) were found to be
nar-rower (FWHM000 = 13 cm−1 and FWHM060 = 13 cm−1) than
for other compositions. It indicates the predominant presence of well-definedenvironmentsbelongingtocrystallinephases, match-ing with
α
-canaphite(FWHMpyro= 10cm−1) forNaPYG-000 andnanocrystallineapatite(FWHMortho=12cm−1)forNaPYG-060.For
the other compositions, broadlines typical of amorphousphases (FWHMpyro = 25–29cm−1 andFWHMortho = 26–29 cm−1) were
observed.Considering the
ν
sPO4 band ofthe orthophosphateion
at 955 cm−1, its intensity was found to increase, as expected, with the increase in the relative proportion of orthophosphate ions inthe initial solution (from NaPYG-010 to NaPYG-050).This trendisopposite forthepyrophosphatestretchingband(
ν
sPO3at
1038 cm−1), showing a decreasein the pyrophosphatewhen the orthophosphate/pyrophosphateratiointhesynthesissolutionwas increased(Table 2 ).
31PMASsolidstateNMRspectrawererecordedinorderto
an-alyze more accurately the environmentsofortho- and pyrophos-phate anions. First, using single-pulse excitation experiments in
NaPYG-000
NaPYG-010
NaPYG-060
Fig. 1.
SEM micrographs of synchesized NaPYG marerials ac dilferenc magnificarions: NaPYG-000 (a. e. i). NaPYG-010 (b. r. j). NaPYG-030 (c. g, k) and NaPYG-060 (d, h. 1).
quantitative mode, the general trends observed in Raman spec
troscopy were confirmed (
Fig.
3
a). Indeed, two regions were ob
served on the spectra, which correspond predominantly to the dif
ferent types of phosphate units, based on previous
NMRstudies
(16
,
17]
and on additional 31P CP-INADEQUATE (discrimination of
pyrophosphate entities) experiments (see Supporting Information,
Sl-4, Fig. Sl ): (i) between 8 and -1 ppm mainly for orthophos
phate species, (ii) between -2 and -10 ppm for pyrophosphate
species
For NaPYG-000 and NaPYG-060, narrow resonances were ob
served, which were respectively attributed to the pyrophosphate
environment of o:-canaphite (-2.5 and -5.8 ppm) and orthophos
phate environment of nanocrystalline apatite (2.8 ppm). However,
it is worth noting that in the case of NaPYG-000, a broad un
derlying component due to pyrophosphate environment was also
observed, accounting for -23% of the overall phosphorus inten
sity. This implies that NaPYG-000 also contains an amorphous
component (in addition to o:-canaphite). For samples NaPYG-010
to NaPYG-050, broad resonances were observed, as expected for
amorphous materials. Their relative intensity was found to vary
in the same way as the proportion of orthophosphate
versuspy
rophosphate species in the initial precursor solution, in fine with
Raman spectroscopie analyses
(
Table 2
).
1H
➔31P CP-MAS experi
ments were also carried out on ail samples (SI-4, Fig. Sl ), showing
that both types of phosphate units are in close proximity to pro
tons, which belong mainly to water molecules (main resonance on
the
1H MAS NMR spectra of ail compounds, centered at -5 ppm,
Fig
.
3
c). A similar observation was made in the case of amorphous
potassium ortho/pyrophosphate materials
(17]
. A more complete
discussion of the
1H and
31P environments is given Iater in this
manuscript.
The local environments of Na
+and Ca
2+cations were also
probed using solid state NMR. Regarding
23Na MAS NMR data, ail
samples were analyied at two different magnetic fields, to ensure a
sounder interpretation of the data, as
23Na is a spin 3/2 quadrupo
Iar nucleus (
Fig
.
3
b, and supporting information SI-5). NaPYG-000
showed two distinct Na environments with well-defined second
order quadrupolar lineshapes (as expected from the crystal struc
ture of o:-canaphite
(36]
, see Fig. S4). However, no broad
23Na res
onance related to the amorphous 31 P was observed, suggesting the
presence of an amorphous calcium pyrophosphate. On the other
hand, NaPYG-060 showed a narrow and yet slightly distributed Na
environment, which corresponds to Na
+-substitutions in nanocrys
talline apatite
(37]
. For samples NaPYG-010 to NaPYG-050, a broad
asymmetric resonance was observed in ail cases, with a tailing to
wards the Iower frequencies. Spectral deconvolutions of these sam
ples were performed considering the presence of two distinct Na
environments (see Sl-5, Fig. S3 and Table S3), as no satisfactory fit
could be achieved using a single one. The relative proportion be
tween both components did not appear to vary significantly with
the ortho/pyrophosphate ratio, and remained equal to -20/80 along
the series. The narrowest
23Na resonance could correspond to Na
+ions which are in a more symmetric and/or more mobile environ
ment due to a higher amount of water molecules in their vicinity.
Conceming
43Ca MAS NMR. ail analyses were performed at
ultra-high field (B
02:. 18.8 T), as
43Ca is a spin 7/2 quadrupo
Iar nucleus of Iow resonance frequency and with a natural abun
dance of only 0.14% (
Fig. 3
-d). Ali
43Ca NMR spectra showed a
broad resonance centered at - 0 ppm. Based on the comparison
of the data recorded at 20.0 and 35.2 T (see SI-6, Fig, S5), and
on previous
43Ca
NMRstudies of amorphous Ca-(pyro)phosphate
phases, the breadth of these signais mainly attests of a chemical
shift distribution, possib(y caused by distributions in Ca-O bond
distance variations
(34
,
38
,
39]
. For two samples (NaPYG-010 and
NaPYG-050), an additional distinct environment was also visible,
with an underlying component at the Iower frequencies
(
Fig. 3
d).
However, due to the difficulties in recording such spectra at natural
abundance, and despite the use of signal-enhancement schemes,
no exact quantification of this component was accessible at this
stage. Taken together,
23Na and
43Ca NMR analyses underscore
the complex nature of the amorphous NaPYG-010 to NaPYG-050
materials, in which not only two different types of anions are
present (ortho and pyrophosphates), but also a variety of cation
environments.
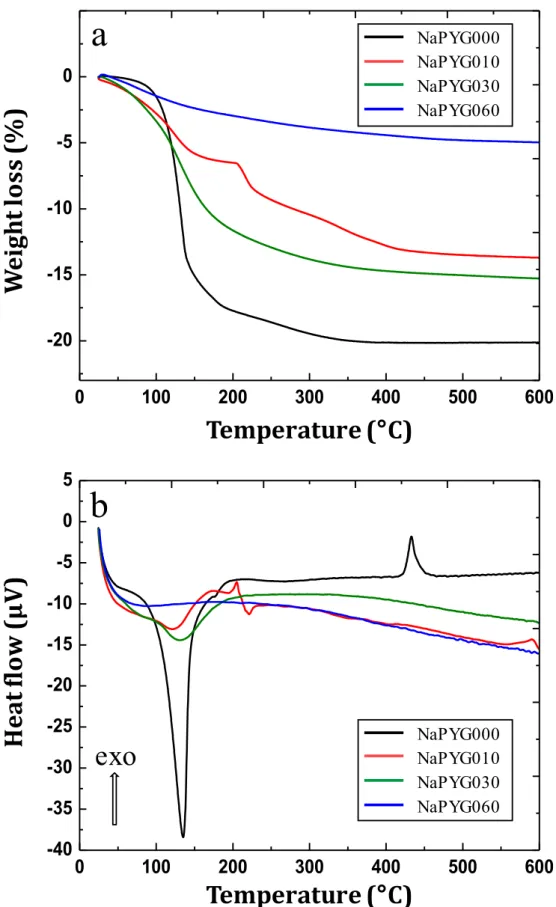
3.3. Thermal behavior and chemical compositions
The thermal evolution of the NaPYG samples was studied by
TGA-DTA.
Fig. 4
shows the TGA and OTA curves of NaPYG-000,
NaPYG-010, NaPYG-030 and NaPYG-060. Only four samples are pre
sented for the ease of reading, since the curves were very simi
Iar for samples NaPYG-020, NaPYG-040 and NaPYG-050 (see Sl-7,
a
� :
il _ . ,
[P yro )/[Ortho]
. .. ! : ! : :� ,. !..
}:\
....
.f...:::
.
/
....
:;_,_})J.}} ___ :__;.;:._ .\
..
=:_;:•
.•
.,.,,. __ ::-_::.)
••
Canaphite
..."
. ... ···• ..-•
... �.• .-···-.. •··�····••'10
20
b
. . .. . ..
.....
·,;;
i::::,_
... . . .. .
30
40
20(
0
)
�
1
...
.
.
.
. .
.
.
. .
.
. . .
. .
.
.
. . . .
. ...
.
.
.
....
.
...
.
50
60
70
.-0;.�
[Pyro]/[Ortho]
..
..
.
. .
.
. .... ...
. ...
-
...
. . .
... .
'...
NaPYG000
NaPYG0l0
NaPYG020
NaPYG030
NaPYG040
NaPYG0S0
NaPYG060
Nano Apatite
Canaphite
NaPYG000
NaPYG0IO
NaPYG020
NaPYG030
NaPYG040
NaPYG0S0
NaPYG060
Nano Apatite
400
600
800
1000
1200
1400
Raman shift (cm-1)
Fig. 2. (a) XRD panerns and (b) Raman specrra of NaPYG materials prepared with dilferent ortho/pyrophosphate molar ratios: from NaPYG-000 to NaPYG-060. Results for the synthesized canaphite and nanocrystalline apatite reference samples are also presented for comparison. (see supporting information).
Fig. S6). The weight Josses of water for all the synthesized ma
terials are reported in
Fig. 4
and summarized in
Table 3
. These
weight Josses can be due to the release of water which is either
adsorbed, directly involved in the bulk structure of these materi
als or resulting from P-OH condensation. For NaPYG-000 (which
contains a significant amount of crystalline o:-canaphite), the main
weight Joss (- 16%) appeared at fairly Jow temperatures (between
80 °C and 170 °C), and was associated with an important endother
mic event. The final weight Joss was below the theoretical value
expected for the Joss of the four water molecules (21.6%) involved
in o:-canaphite (Na
2CaP
20J·4H
20), and consistent with the Joss of
three water molecules. The missing water molecules could be due
to the remaining amorphous component part for which the hy
dration rate can be different from the crystalline part. They could
also be involved in the internai hydrolysis of part of pyrophosphate
ions into orthophosphate ones (
Eq. (1
)
), associated with a small
exothermic contribution around 173 °C (S1-7, Fig. S6). Such a phe
nomenon has been reported in other hydrated calcium pyrophos
phates
(15
,
16)
.
(1)
Finally, the peak at 433 °c corresponds to the crystallization of
anhydrous Na
2CaP
20J phase (XRD data not shown). The general
Fig. 3. (a) 31 P single pulse MAS NMR spectra recorded at 14.1 T, using ν
r = 14 kHz. (b) 23 Na single pulse MAS NMR spectra recorded at 14.1 T using νr = 20 kHz. (c) 1 H single pulse MAS NMR spectra recorded at 14.1 T using νr = 40 kHz. (d) 43 Ca multi-DFS MAS NMR spectra recorded at 18.8 or 20.0 T using νr = 4–6 kHz. Full details on the acquisition conditions are provided in Supporting Information (SI-2).
aspect of the weight loss curves was found to be quite similar
for the amorphous samples NaPYG-020 to NaPYG-050 (see SI-7,
Figure S6), withfinal percentagelosses between12 and 16%. All showedbroadendothermicpeaksattributedtowaterrelease (be-tween50and190°C).Incontrast,afirstplateauwasobservedfor NaPYG-010 (at ~ 194 °C), corresponding to a slowdown ofwater loss.Thisplateauwasprobablyduetoaninternalhydrolysis (asso-ciatedwithanexothermicpeakaround173°C)aswatermolecules areinvolvedinthisreaction.Itisnotobservedfortheother amor-phous samplesprobablydue totheir lower initialpyrophosphate amount.Anendothermiceventassociatedtothisplateauthen oc-curred(from~ 220 °C)duetothequicklossofH2Oafter
hydrol-ysis. ForNaPYG-060(nanocrystallineapatite),theweight losswas continuous(dehydration)upto600°C,withatotalweightloss be-low 5%.Thisvalue isconsistent withthoseofnanocrystalline ap-atites(between4and10%)[40] .
Ca,NaandPamountsinNaPYG-0× 0materialsweremeasured byICP-OESspectrometry,afterdissolutionofthematerials.Results wereusedtoextrapolatethenumberofmolesofeachofthese el-ementsin100gofmaterial(seeTable 3 andsupporting informa-tionSI-8forthe%).UHPLCwasusedtoquantifychlorideionsafter dissolution:forallNaPYGmaterialcompositions,chloride concen-trations were foundto be belowthe detectionlimit, i.e.10−6 mol of Cl− for 100 g of material. For NaPYG-060 the concentrations of Ca, P and Na were consistent with Na-substituted nanocrys-tallineapatite[41] .ForNaPYG-000,thesevalueswerefoundtobe comparable to those ofcanaphite (Ca/P = 0.5and Ca/Na = 0.5): (Ca/P)exp = 0.541and (Ca/Na)exp = 0.548. The difference can be
attributed tothe presence of the amorphousphase asshown by
31P MAS NMR. For the other samples (amorphous), Ca amount
increasedwith theortho/pyrophosphate ratiofromNaPYG-010 to
NaPYG-040 and slightly decreased for NaPYG-050. At the same
time,thephosphorusandsodiumproportionsdecreased.These in-terdependent evolutions ofcalcium, sodium andphosphorus will befurtherdiscussedbelow.
4. Discussion
4.1. TextureandcompositionoftheamorphousNaPYGmaterials
Considering SEM observations (Fig. 1 ) at the lowest mag-nification, the morphologies of the materials containing mixed
ortho/pyrophosphate entities (NaPYG-010 to NaPYG-050) were
similar to those observed for sol-gel derived silica or bioactive silicate glasses [42] . Indeed, without taking any particular pre-caution duringthe gel dryingstep, a powder wasobserved with angularshapes, smooth compact surfaces, andirregular fractures dueto solvent evaporation. This was the casefor all amorphous NaPYG materials, contrary to NaPYG-000 for which the size of powder grains seemed essentially related to the crystallite size of the
α
-canaphite component. Conventional sol-gel processes involvingsiliconalkoxideprecursors[43] ,colloidalgelsareformed (atbasicpH), leadingto materialscomposed eitherofaggregated colloidswhendryingatmoderatetemperatures,orofacontinuous networkof coalesced particles (dependingon the temperatureof the final thermal treatment). Although this conventional sol-gel method was not implemented in the present study, preliminary TEMmeasurementswereperformedonNaPYG-030materials(SI-9, Fig. 7 ),suggestingthatthemillimetricgrainsofamorphousNaPYG materials were not formed of a continuous network but by the aggregationofcolloids/nanoparticles(withadiameterfromafew nanometers to several dozens of nanometers witha hierarchical organization). Considering the large amount of remaining water (between 12 and 16 w%), one could suggest that part of it is dueto non-structuralwater located in the inter-colloidal spaces. Althoughithasnotbedemonstrated,itmayplayakeyroleinthe material cohesion asthe hydrated layer at thesurface of apatite nanocrystals[44] .The relative proportionsof phosphorus atomsinvolved as py-rophosphate (%Ppyro) and orthophosphate (%Portho) species were
Fig. 4. (a) TGA and (b) DTA curves for NaPYG-0 0 0, NaPYG-010, NaPYG-030, NaPYG-060 materials. An expansion of the DTA curves is given in Fig. S6-c.
spectroscopic data (methodology for Raman in SI-3, Table S2). These results (Table 2 ) were compared to the proportions%Ppyro
and%Portho in the initial solutions. First, it should be noted that
similar values were found by 31P NMR formaterials of different
syntheticbatchescorrespondingtothesametargetedcomposition,
showingthereproducibilityofthesesyntheses(SI-10).Second,
Ra-man and NMR quantifications were found to be consistent with
each other (maximum difference of7%).The small variations be-tween both quantifications could be due to slight differences in theRamanscatteringcoefficientsofrespectiveorthophosphateand
Fig. 5. (a) Evolution of weight percentages of Ca 2+ , Na + , PO 43−, P 2 O 74−, H 2 O, and summation of all these ions and (b) Evolution of charges (number of moles of each ionic entity for 100 g of material normalized by its respective charge) for calcium, sodium, total phosphate and summation for each amorphous NaPYG material (x axis is the percentage of P orthophosphate with respect to the total phosphorus content).
pyrophosphate bands (SI-3). The percentages of each phosphate speciesintheamorphoussolidswerefoundtogloballyfollowthe sametrendasinthephosphateprecursorsolutions,i.e.a progres-sive increase of orthophosphates from NaPYG-010 to NaPYG-050, atthe expenseof pyrophosphates(non-linearity ofevolution and excessof orthophosphatesarediscussed below).Combiningthese results withthoseofICP-OES spectrometryandTGA,weight per-centageofCa2+,Na+,PO43−,P2O74− ionsandH2Ohavebeen
cal-culatedforeach composition.Allweightpercentagesarereported in Fig. 5 a (and SI-8, Table S4). The calcium and orthophosphate amountswerefoundtobeclearlycorrelated,andtoincreasefrom
NaPYG-010 to NaPYG-050 for orthophosphates andfrom
NaPYG-010 to NaPYG-040 for Ca2+ions. Simultaneously, sodium and
py-rophosphatecontentsdecreased.
Theionicchargebalancewasdetermined(per100gofsample) foreachcomposition.Asafirstassumption,ortho-and pyrophos-phateionswereconsideredasnon-protonatedinthesecalculations (Fig. 5 b). Theydemonstrate that the relative value of the charge
of phosphate species decreases when the relative amount of
orthophosphates increases. The calcium charge evolution has the opposite curve trend tocompensate it. Calciumis thenthe main counter-ionofphosphatespecies.Itcanbeconsideredasa“linker” maintainingthe cohesionofthephosphatenetwork inthese ma-terials.Sodium,however,appearstoonlyactasapositive“calcium substitute” when the amount of the latter is not high enough to balance the phosphatenegative charges (NaPYG-010 and NaPYG-020).Thiseffectofthelocalelectricfieldshouldbepredominantin absenceofanyadditionalstericeffectasbothcationshavesimilar ionicradius(116 p.m.forNa+ and114pmforCa2+,respectively).
Thesumofchargesisstillslightlynegativeanddecreaseswiththe amountoforthophosphateionsaspreviouslyindicated,suggesting an increaseof protonationofphosphateentities. Consideringthis hypothesis,the calculatedprotonationrateofP-O groupsarelow andvariesfrom 1.0%(NaPYG-010) to 3.6% (NaPYG-050) whatever thephosphatespecies(ortho/pyrophosphate).
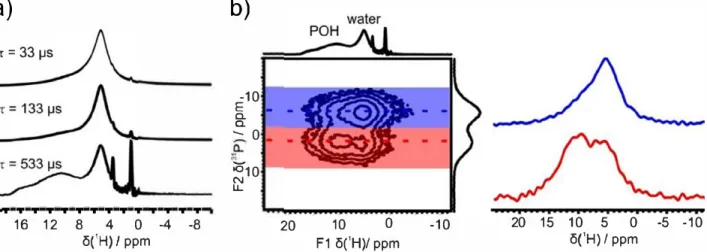
Additional1HsolidstateNMRanalyseswerethuscarriedoutto
investigateinmoredetailtheprotonationofthephosphategroups. Whilesinglepulseexperimentsessentiallyshowedonemain reso-nancecorresponding to watermolecules(Fig. 3 c), Hahnecho ex-periments revealed the presence of underlying signals at higher chemical shifts (> 8 ppm) (Fig. 6 a), which are consistent with protonatedphosphates[45 ,46] .Additional1H→31PHETCOR
experi-mentswereperformed,whichrevealedcorrelationsbetweenthese additional 1H resonances and the ortho and pyrophosphate 31P
peaksatshortcontacttimes,thereby confirmingtheirassignment toP-OHspecies(Fig. 6 bandSI-11,Figs.S9andS10).Pyrophosphate are overall less protonated than orthophosphate. At this stage, a generalchemicalformula(Formula1)canbeproposedby combin-ing31PsolidstateNMR,ICP-OESspectrometryandTGA(assuming
thatthesumoforthophosphateandpyrophosphateequalsto1):
[(Ca2+yNa+zH+3
+x-2y-z)(PO43−)1-x(P2O74−)x](H2O)u (1)
ThestoichiometriccoefficientofH+wasnotmeasuredbut cal-culatedconsideringtheneutralityofthecompound.Inthecaseof NaPYG-030forexample,wedeterminedthatthechemicalformula (Formula2)is:
a)
T= 33 µS
''''''''''''''
b)
-10
E
a.
a.
- 0
!010
N u..16 12 8
4 0 -4 -8
20
10
0
-10
20 15 10 5 0 -5 -10
o(1H) / ppm
F1 oCH)/ ppm
o('H) / ppm
Fig.
6.
(a) 1
H Hahn echo MAS NMR spectra of NaPYG-010 material recorded at 14.1 T, spinning at 22 kHz, using dilferenr echo delays
(r= 33, 133 and 533 µs), showing the
presen�e of protonated pho_sphate resonances
1
at higher chemical shifrs
(>8 ppm): b) 1H➔
31P HETCOR NMR spectrum of NaPYG-010 recorded at 14.1 r, spinning at 22 kHz
and usmg a short contact �me _(0.1 ms). The H Hahn e�ho specrrum r�corded with r = 533 µs is shown on the
1
H projection
ofthe HETCOR. The
1
H NMR spectra in blue
�nd �ed correspond to the H signais of the HETCOR wh1ch correlate w1th the pyro- and ortho-phosphate species, respectively. (For interpreration of the references to colour
m th1s figure legend, the reader 1s referred to the web version of this article.)
80
CU....
�
CU60
e
=
�
40
=
a
0 0 0 N�
0�
p..
�
p..
z
0�
-
z
'
0�
1
p..
t
�
.. z
....
Q, 0 Q,20
0�
1
-0�
C,0
10
20
30
1
0 0�
p..
�
0z
'o::t 0�
�
z
•
Raman quantification
■
NMR quantification
--· Conservation of the ratio
40
50
60
70
10
-
8
-
6
-
4
,_
2
0
80
% p orthophosphate
in the initial solution
Fig.
7. Molar percenrag� of phosphorus atoms involved in orrhophosphate ions
(
compared to pyrophosphate
)
in materials (as detennined
bysolid srate NMR and Raman
spectroscopy
)
as a funct1on of the mo
l
ar percenrage of orthophosphate ions in the initial precursor solution.
In such materials orthophosphates and pyrophosphates can be
considered as forming entities of the network whereas ea
2+are
bridging bivalent cations between phosphate species (pyro- and/or
orthophosphates) and Na
+/H+ non-bridging cations. As stated in
our previous work
(17]
, such compositions are close to those of
invert glasses
(47,48]
, that are elaborated by fusion with high
amounts of glass modifying oxide (CaO) compared to forming ox
ide (P
20
5 ).However, contrary to invert glasses, NaPYG materials
contain a high amount of water and no clear glass transition tem
perature (Tg) was observed in thermal analyses. Hydrated amor
phous calcium pyro- or orthophosphates synthesized at ambient
temperature have also been described
(10
,
15]
, but without both
phosphate entities (or with small amount of orthophosphate gen
erated in
situ(16
])
.
4.2. Contrai of the ortho/pyrophosphate ratio and mechanism of formation of NaPYG materials
The fonnation of amorphous NaPYG materials is allowed by the
inhibitory effect of orthophosphate ions on calcium pyrophosphate
phase crystallization (and reciprocally) as illustrated by the data
(NaPYG crystalline samples resulting from solutions containing
only ortho- or pyrophosphate and amorphous samples resulting
from solutions containing both phosphate species) and already
described in the literature
(48-50]
. The possibility to finely con
trol the pyro/orthophosphate ratio from the initial solution to
give amorphous materials is particularly interesting in a view of
biomedical applications to potentially reach tunable degradation
due to pyrophosphate hydrolysis.
Fig. 7
presents the evolution
of% P orrhophosphate ( vs%P pyrophosphate) in NaPYG materials as a func
tion of the initial%P orrhophosphate in the precursor solution. 31 P
-10
E .5
C.c
0
ëii
5i 0
E
=s
-LL5
10
a
20
15
b
40 20
10
r----1 1 1 1
5
0
�(31P) /ppm
/\
-0 - 2-0 - 4-0 - 6-0
F2 ô{
23Na) / ppm
1 1 1 1 1,_ - - - ..
� E
1-
o.
o.
-0 Q_
IO-5
-10
-15
40 20 0 -20 - 40 - 60
F2 5(
23Na) / ppm
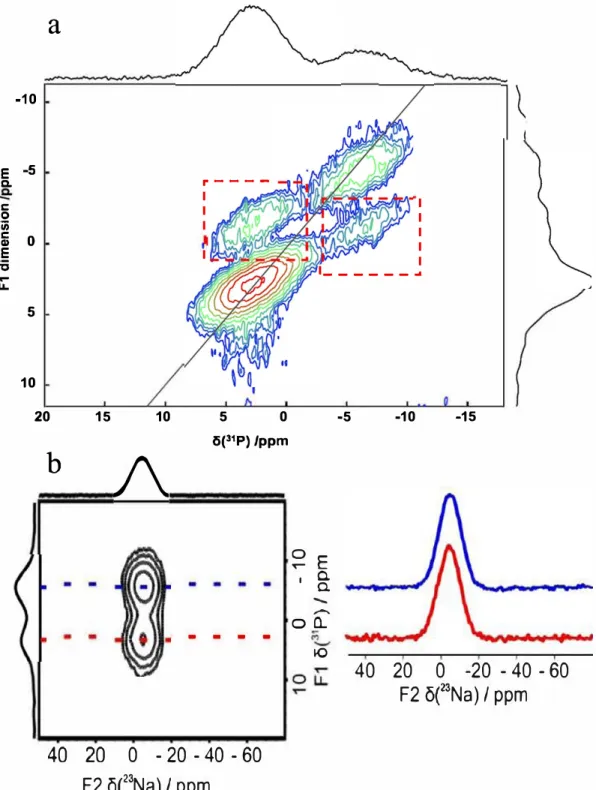
Fig. 8. (a) 31P SQ-DQ speccrum of NaPYG-030 acquired ac 16.4 T, spinning ac 14 kHz. The dashed red boxes show the cross-peaks becween orcho and pyrophosphace unies,
(b) 23Na31 P o-HMQC speccrum of NaPYG-010 macerial, acquired ac 18.8 T using 20 kHz spinning speed (the 23Na NMR speccra in blue and red correspond co correlacions wich
pyro- and orchophosphace ions, respecrively). (For incerprecacion of the references co colour in chis figure legend, the reader is referred co the web version of chis article.)
solid state
NMR
and Raman spectra evolutions showed the same
trend:%P
orchophosphacein the materials are higher than those of
the precursor solution for materials synthesized with solutions
rich in pyrophosphate entities (NaPYG-010/020/030), becoming
doser in the case of orthophosphate rich solutions
(
NaPYG-040/050). Severa( hypotheses can be proposed to explain this
orthophosphates overconcentration in the final material compared
to pyrophosphates.
The first explanation is a partial hydrolysis of pyrophosphates
into orthophosphates for Iow initial ortho/pyrophosphate ratios.
Considering the basic pH of ail the solutions
(
Table 1
), this hydrol
ysis reaction is unlikely to occur d uring the first steps ( colloidal
solution and gel)
(16)
However, a solid state hydrolysis may occur
during the drying step at 70 °C. This assertion is supported by the
fact that stronger correlations are observed on the
1H-
31P HETCOR
spectra between the P-OH groups and the orthophosphate
31P
resonances (rather than the pyrophosphate ones) (see
Fig. 6
b).
Nevertheless, the protonation rates of P-O groups calculated
above (1.0%-3.6%) are too Iow to explain the overconcentration of
orthophosphate.
The second explanation could be linked to the mechanism of colloids formation in the solution and the preferential associa-tionoforthophosphatewithcalcium(vspyrophosphate).Here,this wouldimplytwodifferentpossibilities:(i)orthophosphateand py-rophosphate ions are segregated into two different entities (ions pair,clusters…)andthe solubilityofthe firstone ishigher lead-ingtohigherorthophosphateamountinthefinalmaterial. Unfor-tunately,the pKof such amorphousentities has beenpoorly de-scribed, preventing the (in)validation of this hypothesis, (ii) col-loids are formed of entities associating both pyrophosphate and orthophosphateions,thelatterbeingpredominantduetocharges equilibrium. The results obtained for non-washed samples could confirmthe segregation.Indeed, withoutany washingof thegel, canaphitewasmainlyformed forNaPYG-010, andnanocrystalline
apatite for NaPYG-050, while a mix of both was observed for
NaPYG-030 (SI-12). This is a proof that two kinds of nuclei are formed in the solution either containing orthophosphate or py-rophosphate. Moreover, it demonstrates that the washing step is criticaltoformamorphousmaterials,by removingremainingions (notinvolvedinthenuclei)fromthegel.
Beyond the colloidal solution,one mightwonder ifthe
phos-phate ions are still segregated in amorphous materials and
how they are structured or associated. For this purpose,
amor-phous NaPYG could be compared to amorphous calcium
(or-tho)phosphates (ACPs) [10] for which the most common theory considerstheyareformedby buildingblockscalledPosner’s clus-ter [51] . Theseclusters arearound 1 nm indiameter andhavea corechemicalcomposition ofCa9(PO4)6 andawell-defined
struc-ture [52] . These units have been identified in the first steps of hydroxyapatite and several other crystalline calcium orthophos-phatesformation[53] .Analogousclustermodelsofamorphous Ca-pyrophosphateshavenotbeen describedto date.However, previ-ous PDF (pair distribution function) studies [15 ,54] on hydrated and amorphous pyrophosphate phases (Ca2P2O7.nH2O)
demon-stratedacompletelossofstructuralcoherencebeyondinteratomic separations greater than 7.5–8.0 ˚A that can be attributed to cal-ciumpyrophosphateclusters.
Additional high resolution solid state NMR experiments were performed to analyze in more detail the nature of the domains formingtheNaPYGamorphousmaterials.First,a31PSQ-DQNMR
experimentwasperformed, whichallows probingthe proximities betweenortho- andpyrophosphate units(Fig. 8 -a). The observa-tionof cross peakson the 2D spectrum is a proof ofthe spatial proximitybetweenortho-andpyrophosphateunits.Second,23 Na-31P D-HMQC experiments were performed(Fig. 8 -b), in orderto
probe the Na+/phosphate proximities. The same correlation was observedbetweenthebroad23Naresonanceandthetwotypesof
phosphateunits,whichfurtherconfirmsthattheseanionsare inti-matelyassociatedinthematerial.WhilebothoftheseNMR exper-imentsdemonstratethe existence ofmixedortho-/pyrophosphate entities,theadditionalpresence ofsome segregatedortho- or py-rophosphatedomains/clusterscannotberuledoutatthisstage.
The charge compensation within clusters is meant to be en-suredby both Ca2+ and Na+ ions. A recent molecular dynamics
study [55] has shown that the stoichiometry of orthophosphate clusterscanslightlyvarydependingonseveralparameterssuchas agingofthesolution,protonationofphosphategroups,partial cal-ciumsubstitution bysodiumions,variationofsupersaturation ra-tios.When orthophosphatesare partiallyprotonated,sodium was shown to be a substitute for calcium in the external layer of thePosner-likeclusters, withvariousNa/Ca ratios.Anotherrecent studyhas demonstratedthat sodiumcould facilitatethe aggrega-tionofchargedclustersandcouldsubstituteforcalciumas cluster-bindingcation[56] .Basedonthesestudies,ahypothesis ofmixed ortho/pyrophosphatesclustercanbeproposedexplainingthe23Na
NMRdata(Fig. 3 d).Indeed,thetwoNa+environmentswhichwere
detectedcouldcorrespondtointra-clustersodiumsites(broad sig-nalwhichcorrelates totheorthoandpyrophosphatesignals)and more mobile sodium ions at the surface of theseclusters. Natu-rallysuchhypothesiswillneedtobeconfirmedbycomplementary investigations(coupledWAXS/SAXSassociatedwithsimulation). 5. Conclusion
This article describes the low temperature synthesis of hy-dratedamorphouscalcium/sodiumortho-/pyrophosphatematerials (NaPYG). Forthe firsttime, thesecompounds were obtainedin a largerangeoforthophosphate/pyrophosphatemolarratios,mainly duetoreciprocalinhibitoryeffectofthetwophosphateentitieson theirrespectivecrystallizationwithcalcium.Thisratiocanbe con-trolledinthefinalmaterialasitsevolutionfollowsthesametrend thanthat ofphosphateprecursors intheinitialsolution(with or-thophosphatesinexcessin theamorphousmaterials).Controlling thisratioisofmajorinterestasitcouldopenthewayforthe tun-ingofenzymeorpH-drivendegradationrateofsuchmaterials.
Preliminary results regarding the behavior of the samples in vitro have been obtained, revealing that: (i) the pyrophosphates
could be hydrolyzed in water, in standard SBF solutions and
in TRIS media supplemented or not with ALP enzymes (ii) the
evolutions of the different materials are correlated with their initial ortho/pyro-phosphate ratio. Clearly this ratio appears as a key parameterpotentially offering theabilityto tune upthe bio-logicalbehavior. The complete in vitroand invivo studyofthese materials will be presentedin aforthcoming publication. Beyond their synthesis, multi-scale characterizations have been carried out,includingadvanced multinuclearsolid stateNMR. Thisledto ageneralformulaquantifying theircomposition.The datasuggest that thesesolids are formed by theaggregation ofcolloids. They are also consistent with a cluster-based material. Although their existencewillneedtobeinvestigatedindetailby complementary techniques, such clusters could be formed by calcium, sodium and partially protonated ortho- and pyrophosphate entities and beassociated togetherthroughsurfacewaterandsodiumcations, ensuringtheoverallcohesionofthewholematerial.
DeclarationofCompetingInterest
Theauthorsdeclarethattheyhavenoknowncompeting finan-cialinterestsorpersonalrelationshipsthatcouldhaveappearedto influencetheworkreportedinthispaper.
Acknowledgments
The authors would like to thank the Agence Nationale de la Recherche(PyVerresproject-grantn°ANR-16-CE19-0013)for sup-portingthisresearch work. Theauthors are gratefulforaccessto UK 850MHz solid-state NMR Facility used in this research and fundedbyEPSRC andBBSRC (contractreferencePR140003),aswell asUniversity of Warwick includingviapartfundingthrough Birm-inghamScienceCityAdvancedMaterialsProject1and2,supported by Advantage West Midlands (AWM)and the European Regional Development Fund (ERDF).Aportion ofthiswork wasperformed attheNationalHighMagneticFieldLaboratory,whichissupported by the National Science Foundation Cooperative Agreement No. DMR-1157490 &DMR-1644779 ,andtheStateofFlorida.TheFrench RégionIledeFrance-SESAMEprogramisacknowledgedfor finan-cial support (700MHz NMR spectrometer). DinuIuga is acknowl-edgedfortheirassistanceinsomeoftheNMRexperiments.
Supplementarymaterials
Supplementary material associated with this article can be found,intheonlineversion,atdoi:10.1016/j.actbio.2019.12.027 .