Malleable polymers capable of stimuli-controlled shape memory, self-healing and motion
189
0
0
Texte intégral
Figure
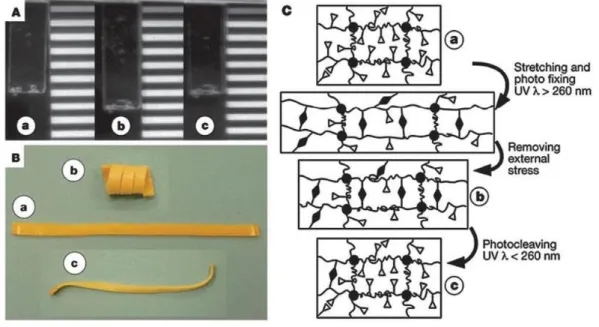

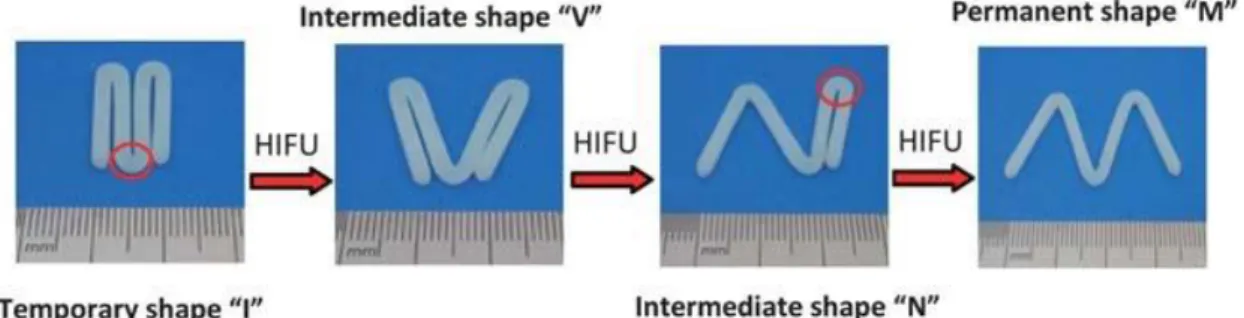
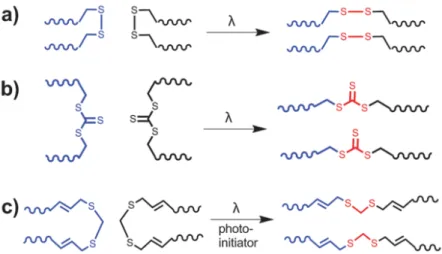
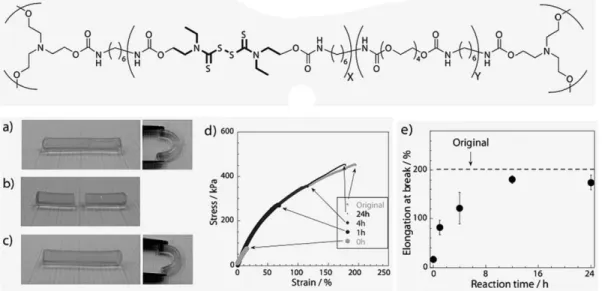
+7

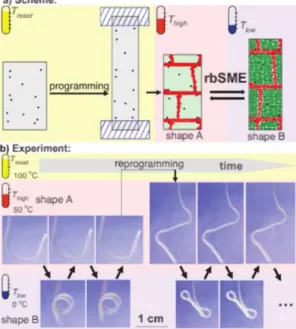
![Figure 13. Four main classes of polymer actuators based on different actuation mechanisms [90]:](https://thumb-eu.123doks.com/thumbv2/123doknet/3537463.103583/55.918.298.619.446.755/figure-classes-polymer-actuators-based-different-actuation-mechanisms.webp)

![Figure 20. Digital images of the twisted films under different humidity conditions. [133]](https://thumb-eu.123doks.com/thumbv2/123doknet/3537463.103583/63.918.342.577.210.448/figure-digital-images-twisted-films-different-humidity-conditions.webp)
Documents relatifs