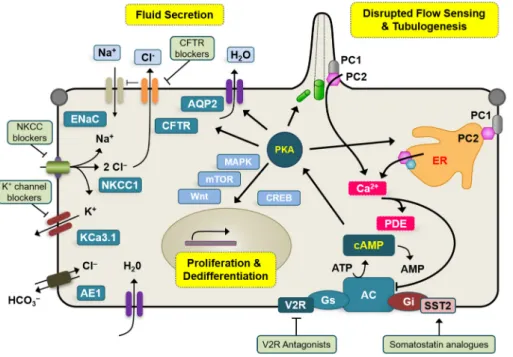
Targeting chloride transport in autosomal dominant polycystic kidney disease.
Texte intégral
Figure
Documents relatifs
After introducing some properties of the theory of ordinary differential equations, we provide a rigorous computational method for finding the periodic solution
Neptunium sorption and redox speciation at the illite surface under highly saline conditions.. Nidhu Lal Banik, Rémi Marsac, Johannes Lützenkirchen, Christian Maquardt, Kathy
In this study, the shell-shaped model was fit to the g-value data to determine CME speeds in the solar wind, and the radial variation of propagation speeds between the corona and
Allez donc chercher la vérité dans cet imbroglio où, depuis toujours, tous avancent avec dans une main le Livre saint qui prône l’amour du prochain et dans
Toutes les salles sont équipées des technologies les plus récentes avec, pour les deux plus grandes, l’image en 3D et, surtout, dans la salle N°1, le nouveau son 3D Dolby Atmos
Cela rend également visible ce qui, pour des raisons de sécurité, se déroulera dans des halles fermées – de l’excavation des déchets par un système à ponts
(collective noun + singular or plural verb; audience can also be used as a countable noun with a normal plural: audiences are; also: class, club, committee, company,
Nous$ avons$ pour$ l'instant$ évoqué$ uniquement$ l'huile$ de$ palme$ contenue$ dans$ les$
