HAL Id: dumas-01681472
https://dumas.ccsd.cnrs.fr/dumas-01681472
Submitted on 11 Jan 2018HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Conception d’un système de veille et de consolidation
des développements cliniques avancés en matière de
thérapie génique
Lionel Cadot
To cite this version:
Lionel Cadot. Conception d’un système de veille et de consolidation des développements cliniques avancés en matière de thérapie génique . Génie des procédés. 2015. �dumas-01681472�
CONSERVATOIRE NATIONAL DES ARTS ET METIERS
PARIS
MEMOIRE
présenté en vue d’obtenir
le DIPLOME D’INGENIEUR CNAM
SPECIALITE : GENIE DES PROCEDES
PARCOURS : PROCEDES PHARMACEUTIQUES
par
CADOT, Lionel
CONCEPTION D’UN SYSTEME DE VEILLE ET DE CONSOLIDATION DES DEVELOPPEMENTS CLINIQUES AVANCES EN MATIERE DE THERAPIE GENIQUE
Soutenu le 14 janvier 2015
Jury :
- Président : Pr Jean-Louis HAVET – Professeur des universités – CASER1
Cnam GPIP Membres académiques :
- Dr Serge STAINEMESSE - Maître de conférences – CASER Cnam-GPIP : Tuteur
- Dr Marie DEBACQ - Maître de conférences – CASER Cnam-GPIP
Membres représentant l’entreprise – Astellas France :
- Dr Eric CHAUVEAU - Directeur Médical – Département Médical (MED)
- Maryse CRABOS - Responsable des Opérations Cliniques – MED
- Laurence BEAUVY - Directeur Formation et Développement RH - Département RH
1
Remerciements
En entamant mon cursus au conservatoire national des arts et métiers, je n’imaginais pas alors que cet acte allait être si déterminant pour les 10 années et plus qui suivirent.
J’ai d’abord une pensée pour mes professeurs du CNAM, pour leur enthousiasme, leur passion, et leur soif de transmettre leur savoir. Aucun ne m’a laissé indifférent. Tous dans leur style propre étaient animés par une volonté commune de donner du sens à leur discipline, en en montrant la beauté à leurs élèves, tout en la mettant en perspective du monde qui nous entoure. Les convictions techniques et scientifiques étaient affichées mais toujours argumentées et discutées dans un esprit d’ouverture. Leur enseignement est l’un des meilleurs qu’il m’ait été donné de recevoir. Depuis des années déjà il accompagne régulièrement mes décisions en entreprise, m’a ouvert des opportunités insoupçonnées et je m’y référerai encore régulièrement dans le futur. A tous, pour tout cela je vous remercie.
J’ai une pensée toute particulière pour Mr Bertrand Roudier, Mme Renée de Challemaison et Mr Serge Stainemesse dont les noms risquent bien de ne jamais sortir de ma mémoire tant leur investissement à me former, à nous former fut sans faille.
Je tiens également à exprimer ma sincère reconnaissance aux managers qui m’ont fait confiance et soutenu dans cette démarche chez GSK, Genzyme et maintenant Astellas. Je veux parler d’xxxxxxx, xxxxxx xxxxxxx, xxxxxxxxx xxxxxx et xxxx xxxxxxx. Ils m’ont permis de me rendre aux cours du soir, d’intégrer ce projet dans mes objectifs, de disposer d’un appui logistique bibliographique lorsque nécessaire, et enfin de m’accompagner dans cette étape finale.
Parvenir à la dernière étape de cet objectif personnel ne fut pas toujours un long fleuve tranquille, ou peut-être seulement un peu, au tout début ce qui coïncide également avec le démarrage de ma carrière professionnelle dans l’industrie pharmaceutique et le moment où je quittais le sud de la France pour rejoindre ma future femme. Avec le temps, les responsabilités professionnelles augmentant, une équipe à manager, des règles européennes nouvelles faisant évoluer les formations, les responsabilités personnelles s’étoffant également, un logement à rénover, une enfant, puis deux, un déménagement,…le temps est devenu de plus en plus rare, précieux. J’ai lutté, j’ai douté, j’ai négocié, je me suis sans cesse réorganisé pour ne pas abandonner mais aujourd’hui, je peux affirmer que je n’ai pas de regret. Ces moments, ces étapes je n’aurai pu les franchir sans mes proches. Ils ont non seulement accepté mes périodes de travail, de révisions, de préparation dans des moments familiaux, parfois festifs ou de rassemblement qui ne s’y prêtaient guère et ont eu toujours l’élégance de ne pas me les reprocher et même souvent de me les faciliter. Je pense et remercie donc ma famille, parents, sœur, belle famille, oncles, tantes, amis, proches pour ces moments qu’ils ont acceptés et compris.
A mes deux filles, xxxxxxxx x ans et xxxxx xx mois, je présente mes excuses pour certains moments d’indisponibilité ces dernières années et j’espère que plus tard vous serez fières de mon engagement.
Enfin, je ne peux clore ce chapitre sans rendre un hommage tout particulier à celle qui m’accompagne dans cette aventure depuis plus de 10 ans, ma femme. Sans sa patience à mon égard, sa compréhension de ma démarche personnelle, nos discussions, je ne serai pas là aujourd’hui. xxxxxxxx, merci !
Table des matières
ACRONYMES……….……..6
1. INTRODUCTION ... 7
1.1. Définitions et concepts de la thérapie génique ... 7
1.1.1. Gène Médicament / Microchirurgie génique 7
1.1.2. Thérapie génique somatique / Germinale 8
1.1.3. Administration du transgène In vivo ou Ex vivo 11
1.2. Des balbutiements aux premiers succès ... 11
2. METHODOLOGIE EMPLOYEE ... 16
2.1. Périmètre ... 16
2.2. Matériel et Méthodes ... 17
2.2.1. Analyse de l’existant 17
2.2.2. Sources d’informations de référence 17
2.2.3. Procédure suivie 19
3. QUALITE DE LA BASE DE DONNEES ... 24
3.1. Eléments d’identification et de méthodologie des essais ... 24
3.2. Résultats des essais ... 25
4. RESULTATS ... 26
4.1. Résultats portant sur les essais cliniques ... 26
4.1.1. Distribution quantitative et qualitative par attributs (hors axes de recherches et indications) 26
4.1.1.1. Distribution par Phase et Status ... 26
4.1.1.2. Distribution par zone de réalisation des essais ... 27
4.1.1.3. Distribution par promoteurs des essais ... 28
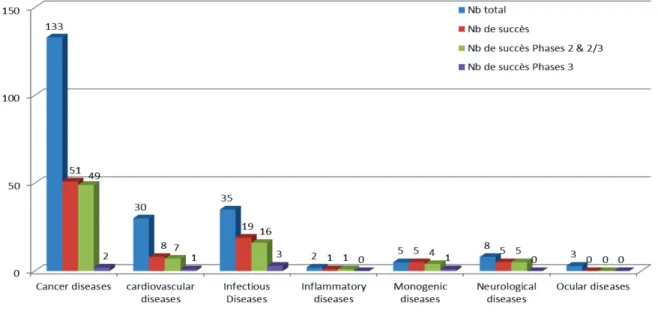
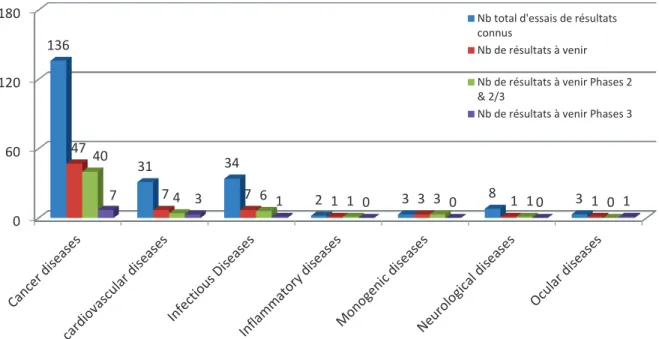
4.1.2. Distribution quantitative et qualitative par axes de recherches et indications 28 4.1.2.1. Distribution par axes de recherche ... 28
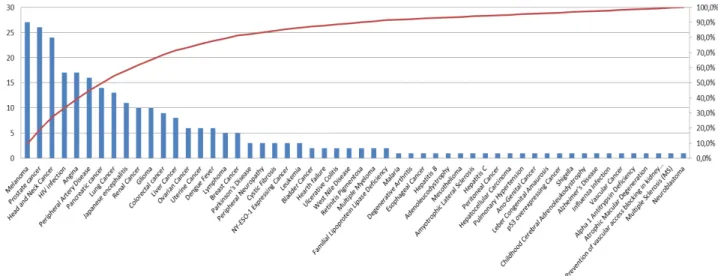
4.1.2.2. Distribution par indications ... 32
4.1.3. Distribution quantitative et qualitative en fonction des gènes étudiés 40
4.1.3.1. Distribution par type de gène des essais ... 40
4.1.3.2. Distribution des essais en fonction des gènes ... 41
4.1.4. Distribution quantitative et qualitative en fonction des vecteurs étudiés 44
4.2. Méthode d’identification des produits à potentiel de succès ... 46
5. CONCLUSION... 55
ANNEXES ... 588
Annexe I : sites internet principaux d’informations sur les essais cliniques ... 58
Annexe II : Spécifications des alertes bibliographiques mise en œuvre ... 59
Annexe III : FICHE PRODUIT ... 60
Annexe IV : Tableaux non intégrés dans le corps du rapport ... 67
Annexe V : Fiche signalétique de l’étude US-1163 telle qu’elle est visualisable dans la base de données ... 82
Annexe VI : Synthèse du travail réalisé en tant qu’élève ingénieur ... 83
ACRONYMES
AAV : Adeno-associated virus ADN : Acide désoxyribonucléîque ARN : Acide ribonucléîque
CGM (Cellules Génétiquement Modifiées)
Del-1 : Developmentally regulated endothelial locus-1 FGF : Fibroblaste Growth Factor
HGF : Hepatocyte Growth Factor HIF-1α : Hypoxia Inducible Factor IL2 : Interleukine 2
MTG : Médicament de Thérapie Génique NIH: National Institute of Health
PDGF : Platelet Derived Growth Factor PTG : Produit de Thérapie Génique
VEGF : Vascular Endothelial Growth Factor
NOTE EXPLICATIVE CONCERNANT LES TABLEAUX, GRAPHIQUES ET SCHEMAS :
Les tableaux et graphiques présentés dans ce document sont issus pour la plupart du système présenté. Ce système est basé actuellement sur l’application Microsoft Excel version Française (Microsoft Office Professionnel 2010). Pour cette raison, les champs issus de l’application Microsoft elle-même, notamment ceux d’affichage des résultats des tableaux croisés dynamiques (Etiquettes de colonnes, Nombre de, Total général…) apparaissent en français alors que les contenus et libellés des variables ont été saisis en anglais, en vue de faciliter les échanges externes ultérieurs. Cette note s’applique aussi bien au corps du texte qu’aux annexes.
Dans le corps du texte, les tableaux créés en dehors de l’application sont en revanche en Français. Les Schémas sont dans la langue d’origine du document dont ils sont extraits.
1. INTRODUCTION
1.1. Définitions et concepts de la thérapie génique
1.1.1. Gène Médicament / Microchirurgie génique
Le terme de thérapie génique (TG) peut recouvrir 2 types de techniques thérapeutiques
aujourd’hui : l’utilisation de gènes comme médicaments et la réparation des gènes1. Le second
procédé, celui d’une microchirurgie génique, correspond probablement à ce à quoi pouvaient penser les généticiens il y a quelques décennies lorsqu’ils imaginaient que les maladies géniques pourraient un jour être traitées et radicalement guéries. Cependant ce n’est que récemment que des succès expérimentaux de corrections de gènes in situ par l’utilisation de chimères d’oligonucléotides ADN / ARN ont permis d’envisager qu’un tel rêve puisse devenir réalisable dans des maladies génétiques avec mutations ponctuelles des allèles concernés. En revanche, ces résultats in vitro n’ont jamais pu être confirmés in vivo et ne restent pour l’instant qu’une piste
de recherche2, qui semble délaissée aujourd’hui au profit de la technologie des méga nucléases
développée par la société Cellectis (www.cellectis.com). L’essentiel des travaux et des essais réalisés à ce jour concerne pour l’essentiel la première acceptation de l’expression « thérapie génique » soit la notion de gène médicament. Il s’agit d’une évolution singulière du statut des gènes, ceux-ci dépositaires de parcelles du programme génétique, sont en effet responsables de maladies qualifiées de maladies génétiques dénombrées aujourd’hui à plus de 4000 qu’il s’agisse de maladies monogéniques, multifactorielles (plusieurs gènes), d’anomalies chromosomiques
ou de mutations dans l’ADN mitochondrial 3.
La thérapie génique pourra s’appliquer évidemment aux maladies génétiques, affections héréditaires ou acquises qui peuvent bénéficier du traitement par une protéine recombinante. En effet, il est en principe toujours possible de remplacer la protéine de substitution par le gène qui va en commander la synthèse dans les cellules du malade qu’il faut soigner. En ce sens la thérapie génique constitue la troisième étape de l’utilisation des protéines médicamenteuses après la purification associée parfois à l’humanisation chimique, (par exemple à partir de matériel biologique animal) et après l’utilisation du génie génétique (où les microorganismes sont utilisés pour la production). La thérapie génique, dans son approche la plus directe, transfère le gène d’intérêt dans les cellules de l’organisme qu’il faut soigner. Ces cellules deviennent alors les micro-usines de fabrication du gène mais également les systèmes de délivrance du médicament
au malade1.
Plus officiellement, l’ANSM (agence nationale de sécurité du médicament et des produits de santé) définit un médicament de thérapie génique (MTG) au 28/09/2012 sur son site internet comme tout produit obtenu par un ensemble de procédés de fabrication visant au transfert d’un gène prophylactique, diagnostique ou thérapeutique chez l’homme et son expression consécutive in vivo. Toujours selon l’ANSM, le transfert d’un gène implique un système d’expression contenu lui-même dans un système d’administration appelé vecteur qui peut être d’origine virale ou non. Les MTG étudiés dans ce projet de fin d’étude répondent à cette définition. Même si considérée par certains auteurs comme de la thérapie génique, la thérapie dite « antisens » qui consiste à neutraliser une séquence de mRNA par le DNA complémentaire ainsi que toutes les techniques dérivées de cette approche n’ont pas été incluses dans le périmètre de ce travail.
1.1.2. Thérapie génique somatique / Germinale
Les principes généraux concernant la thérapie génique germinale évoqués ci-dessous sont issus ou inspirés du livre d’Axel Kahn « Thérapie Génique – l’ADN MEDICAMENT » de 1993 édité par John Libbey Eurotext. Ils ont été actualisés avec quelques références plus récentes citées dans le texte mais les principes fondamentaux restent pour la plupart toujours d’actualité.
Il existe théoriquement deux moyens de pratiquer une thérapie génique germinale. La première consiste à introduire la modification dans toutes les cellules d’un embryon très précoce, voire dans l’unique cellule, de l’œuf nouvellement fécondé, issu de la fusion des gamètes mâle et femelle après fécondation. Dans ce cas, la modification se retrouvera dans toutes les cellules de l’organisme développé, y compris dans les gamètes, et sera donc transmise à la descendance comme un nouveau caractère génétique. Chez l’animal, cette approche, dénommée transgénèse, est maintenant très couramment pratiquée ; elle permet d’obtenir des modèles animaux de maladies humaines pour la recherche, de créer des animaux produisant des substances d’intérêt biologique, ou est utilisée pour étudier le rôle ou le fonctionnement de certains gènes. La transgénèse ajoute un gène au patrimoine génétique d’un organisme, en l’insérant soit au hasard dans les chromosomes, soit à sa place normale, en remplacement du gène dont l’altération est responsable de la maladie. On parle dans ce dernier cas de recombinaison homologue, applicable à l’animal mais non, dans l’état actuel de la science, à l’homme, au moins dans le cadre d’une modification génétique de l’embryon entier. En revanche, la transgénèse avec intégration au hasard serait, pratiquement et théoriquement possible chez l’homme.
Une deuxième forme de TG germinale consisterait, dans un organisme constitué, à intégrer un gène spécifiquement dans les cellules germinales. Il n’existe jusqu’à présent, aucun moyen de parvenir à un tel résultat. Ainsi la seule thérapie génique germinale qui pourrait être appliquée à l’homme est la transgénèse par apport d’un nouveau gène dans l’embryon précoce humain ; il aboutirait à un organisme dont les cellules somatiques seraient génétiquement modifiées et qui transmettrait par ses cellules germinales cette modification à une partie de sa descendance. Une telle méthode n’a pas pour l’instant d’indication thérapeutique chez l’homme. Pour réaliser une telle expérience de transgénèse, il faudrait disposer d’embryons unicellulaires comme c’est le cas dans la fécondation in vitro, où ils sont obtenus en plusieurs exemplaires ou par lavage utérin après induction d’une superovulation et fécondation naturelle. Dans les deux cas, la fécondation à partir des gamètes des deux parents à risque, aboutit à un mélange d’embryons porteurs de la tare et (ou) malades, et d’embryons sains. Raisonnablement, il n’est pas envisageable de tenter de modifier au hasard un des embryons sans savoir auparavant s’il est sain ou atteint. Il serait donc nécessaire, pour les couples risquant d’avoir un enfant atteint, de faire un diagnostic pré- -implantatoire, c’est-à-dire avant la réimplantation de l’embryon dans les voies génitales de la femme pour distinguer les embryons atteints des embryons normaux. Cela conduirait à écarter les embryons atteints et à ne transférer dans l’utérus de la mère que les embryons n’ayant pas la tare pour éviter la naissance d’un enfant malade. Axel Kahn a d’ailleurs écrit : « Celui qui écarterait l’embryon normal pour tenter de modifier, avec des résultats incertains, et de toute façon inconstants, l’embryon atteint, serait un fou dangereux surtout après avoir identifié que d’autres zygotes ne sont pas atteints ».
Ainsi, le diagnostic sur l’embryon très précoce d’une maladie génétique, prélude théorique indispensable à une thérapie génique germinale, aboutit en réalité à un tri d’embryons et non pas à une thérapie génique, ce qui pose de nombreuses questions philosophiques, éthiques et morales. Ce type d’approche relève en effet finalement plus de « l’amélioration génétique de l’espèce » (genetic enhancement) que du traitement germinal d’une maladie génétique. En dehors même de cet aspect du problème, les indications potentielles d’une telle TG ne sont pas évidentes en l’état de la science. Sa justification pourrait être d’éviter que l’individu traité ne transmettre la maladie à ses descendants. Or, de toute façon, vu que la TG génique s’effectue en l’état de la science au hasard, la probabilité que la tare soit complètement corrigée par l’intégration du gène au hasard est quasi nulle. Ainsi, indubitablement 50% des gamètes d’un individu issu d’un embryon ayant subi un tel traitement continueront de transmettre la tare. En réalité, il n’existe que deux cas où l’intervention sur les cellules germinales humaines pourrait avoir une indication thérapeutique. Le premier est celui d’une maladie héréditaire qui frapperait primitivement les cellules intervenant dans la gamétogénèse (formes exceptionnelles de stérilité génétique par anomalie fonctionnelle des gamètes). Le second est celui des individus homozygotes pour des maladies dominantes désirant avoir des enfants (par exemple, des sujets atteints de chorée de Huntington, de polykystose rénale héréditaire dominante, chez lesquels les exceptionnels homozygotes pourraient n’être pas beaucoup plus gravement atteints que les hétérozygotes).
En France, ces pratiques sont interdites par la loi de Bioéthique dont la dernière modification
date d’août 20134. La loi dispose désormais qu'aucune recherche sur l'embryon humain ni sur les
cellules souches embryonnaires ne peut être entreprise sans autorisation. Cette autorisation sous la responsabilité de l’Agence de Biomédecine est assortie de certaines conditions: "la pertinence scientifique de la recherche", et la "finalité médicale" de cette recherche notamment. Le texte précise en outre qu'une recherche ne peut être menée qu'à partir d'embryons surnuméraires conçus in vitro dans le cadre d'une assistance médicale à la procréation et qui, soit ne font plus l'objet d'un projet parental soit que les parents choisissent de donner à la recherche. On aurait donc pu penser qu’une transgénèse pourrait être autorisée dans ce cadre. Il n’en est rien, l’agence de biomédecine sur son site internet rappelle et renforce un des principes issus de la loi de 2011 selon lequel la création d’embryons transgéniques ou chimériques est interdite. Autre pilier de la loi de bioéthique institué par le texte de juillet 1994, « tout pratique eugénique tendant à l’organisation de la sélection des personnes est interdite » et « sans préjudice des recherches tendant à la prévention et au traitement des maladies génétiques, aucune transformation ne peut être apportée aux caractères génétiques dans le but de modifier la descendance de la personne ».
Aux USA néanmoins, il n'existe aucune réglementation limitant la thérapie génétique germinale ou l'utilisation d'embryons humains pour autant que ces techniques obtiennent l'autorisation de la FDA. Pour l’instant, même si le débat est régulièrement relancé aux USA par les partisans de cette approche, la FDA n’a jamais autorisé de recherche de Thérapie génique germinale. Par ailleurs, je n’ai pas connaissance à la date de ce rapport du moindre essai clinique chez l’homme avec cette approche, unanimement condamnée d’un point de vue éthique.
La thérapie génique somatique se rapproche plus de la thérapeutique commune car il s’agit de traiter la maladie par un médicament, le gène, ou de corriger la mutation d’un gène dans le tissu atteint sans influer sur l’hérédité du malade. Les seuls essais de thérapie génique conduits chez l’homme jusqu’à mi 2013 concernent la thérapie génique somatique.
Alors, existe-t-il une voie de TG alternative à la TG somatique ? Cette voie pourrait bien être celle des cellules pluripotentes induites (iPS). Les iPS sont des cellules adultes spécialisées provenant de pratiquement n’importe quel type cellulaire que l’on reprogramme ou déprogramme génétiquement pour les rendre pluripotentes, c’est-à-dire capables de se multiplier à l’infini et de se différencier en types de cellules qui composent un organisme adulte, exactement comme une cellule souche embryonnaire. Cette découverte a valu le prix Nobel de médecine 2012 à Shinya Yamanaka, le chercheur japonais qui a mis au point la technique six ans plus tôt à l’université de Kobé. Les cellules iPS ont les mêmes atouts que les cellules souches embryonnaires humaines mais « elles ont des avantages supplémentaires : elles sont faciles
d’accès, par simple biopsie chez l’adulte, et leur utilisation ne pose pas de problème éthique »5.
Elles sont déjà utilisées pour créer des modèles de maladies génétiques et portent de grands espoirs dans la médecine régénérative, notamment dans les maladies neurodégénératives ou les cellules souches adultes sont très complexes à obtenir.
Les autorités sanitaires du Japon viennent de donner le feu vert aux chercheurs pour qu’ils procèdent aux premiers essais cliniques de cellules souches pluripotentes induites sur l’homme. Pour cette première mondiale, les tests porteront sur une atteinte de l’œil, la dégénérescence
maculaire liée à l’âge (DMLA)6. L’enjeu est de taille puisque cette maladie est la première cause
de cécité des plus de 55 ans dans les pays industrialisés. Une fois implantées dans la rétine, il est espéré que les iPS régénèrent l’œil malade. Une réserve s’impose cependant. Les cellules souches embryonnaires demeurent des cellules physiologiques « naturelles ». La reprogrammation des cellules iPS pose en effet la question d’éventuelles mutations qui pourraient entraver leur fonctionnement. Ce premier essai au Japon devrait apporter des réponses à cette question. Il ne s’agit pas dans cet essai d’une thérapie génique mais d’une thérapie cellulaire d’un genre nouveau. En revanche, il n’y a aucun frein méthodologique ou éthique à ce que ces cellules IPS puissent être dans le futur modifiées génétiquement, multipliées et réinjectées à l’homme ouvrant ainsi une nouvelle voie de la thérapie génique.
D’ailleurs, dans le cadre d’un projet mené par des équipes de l’université de Cambridge et du Sanger Institute, en collaboration avec une équipe de l’Institut Pasteur et de l’Inserm, des chercheurs ont montré pour la première fois que des cellules iPS, produites à partir de cellules de patients atteints d’une maladie du foie (mutation ponctuelle dans le gène de l’enzyme alpha-1-antitrypsine), peuvent être génétiquement corrigées puis différenciées en cellules hépatiques pour participer à une régénération du foie dans un modèle animal de la maladie. Ces travaux,
publiés le 12 octobre 2011 sur le site de la revue Nature7, constituent une preuve de concept
majeure pour envisager le recours futur à ces cellules souches autologues chez l’Homme, en vue d’une thérapie génique.
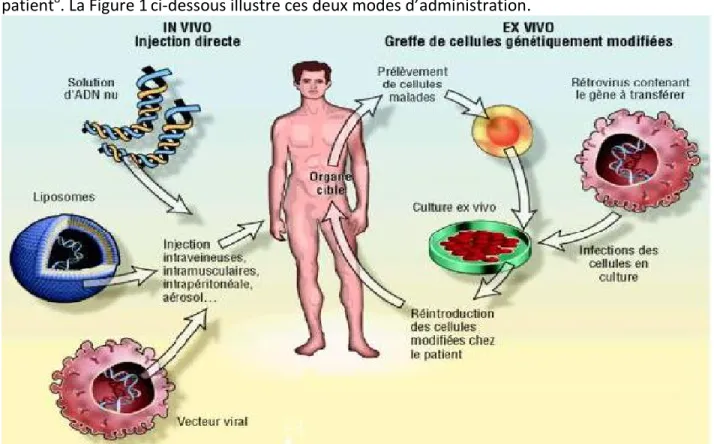
1.1.3. Administration du transgène In vivo ou Ex vivo
Deux modes d’administration des produits de thérapie génique (PTG) sont communément utilisés chez l’Homme :
- l’administration dite in vivo - l’administration dite ex vivo
L’administration in vivo consiste à injecter directement, au niveau de l’organe, du tissu cible ou même dans la circulation générale, le PTG alors que l’administration ex vivo consiste à transfecter in vitro des cellules (autologues, allogéniques ou xénogéniques) avec un vecteur portant le gène d’intérêt, puis à implanter ou réimplanter ces cellules génétiquement modifiées (CGM) chez le
patient8. La Figure 1ci-dessous illustre ces deux modes d’administration.
Figure 1 : modes d’administration chez l’homme des PTG
1.2. Des balbutiements aux premiers succès
Les premiers travaux entre les années 70 et 90 consacrés à la thérapie génique ont été réalisés dans des conditions scientifiques, sinon éthiques que l’on peut qualifier de hasardeuses :
- en 1973, l'injection par Rogers et al. du virus du papillome de Shope à des filles souffrant d'hyperargininémie, dans l'espoir que cela permettrait le transfert d'un gène viral
imaginaire d'arginase, ne devait évidemment donner aucun résultat1.
- En 1980, la transfection du gène de P-globine dans des cellules médullaires de jeunes thalassémiques, expérience qui reposait sur des résultats expérimentaux antérieurement obtenus par Martin Cline et al. chez la souris, devait également aboutir à un échec total...
- En 1983, un congrès scientifique (Banbury Meeting à Cold Spring Harbor Laboratory), dans le Long Island aux États-Unis était pour la première fois consacré à la thérapie génique des
maladies génétiques1.
- le 22 mai 19899
premier transfert d'un gène marqueur dans les lymphocytes infiltrant les tumeurs, sous l'égide de Stephen Rosenberg et al, qui devait marquer la naissance officielle de la thérapie génique « légale ».
Ces premières tentatives furent très critiquées principalement en raison d’un manque de recul sur les techniques et du manque de données pertinentes sur l’animal avant le passage à l’homme.
À partir de 1991, les essais cliniques de TG chez l'homme se multiplient, consacrés pour près des deux tiers aux cancers, alors que les essais thérapeutiques pour des maladies génétiques héréditaires restent limités aux déficits immunitaires, à l'hypercholestérolémie familiale et à la mucoviscidose. Très rapidement, les maladies génétiques apparurent en effet beaucoup moins propices à ce type de traitement, en l’état de la science, que d'autres affections acquises ne nécessitant pas obligatoirement une correction très prolongée de cellules parfois nombreuses et difficiles à atteindre.
En 1995, un directeur du NIH américain, Harold Varinus va missionner une commission d'experts dirigée par Arno Motulski et Stuart Orkin, pour dresser un bilan critique des tentatives de Thérapie Génique sur les 5 années précédentes :
- 130 essais cliniques menés en 5 ans, conduits avant tout aux États-Unis, - plus de 1000 malades ayant reçu un PTG
Le constat rendu public en 1995 conclue sur de nombreuses difficultés :
- performances insuffisantes des vecteurs et nécessité d'améliorer les modes de délivrance (besoin d'efforts supplémentaires des laboratoires de recherche dans ce domaine). Cela a permis d’ouvrir la voie et d’identifier où les efforts devaient se porter. La plupart des équipes vont à partir de là focaliser leurs efforts sur le développement de nouveaux vecteurs, permettant d'espérer une expression ciblée (tropisme), contrôlée et prolongée des transgènes thérapeutiques. Aujourd’hui la Vectorologie demeure au cœur des recherches menées dans ce domaine et représente l’un des enjeux majeurs pour les succès futurs de l’approche.
- Développement croissant des MTG comme moyen de délivrer une protéine médicamenteuse quelconque dans des maladies fréquentes mais multifactorielles. Cette tendance est liée principalement à l'importance du marché de ces affections, comparée à celui de la plupart des maladies génétiques, et donc à un retour sur investissement plus avantageux en cas de succès.
Pendant les 15 années qui suivent :
- les recherches en TG autour des désordres héréditaires se poursuivent malgré tout, stimulées notamment par de puissantes associations de malades dont l'Association française de lutte contre les myopathies en France (AFM).
- le traitement par les gènes fait la preuve de son efficacité dans de nombreux modèles animaux de maladies.
- de nouvelles générations de vecteurs, viraux et non viraux, apparaissent grâce aux avancées des recherches en Vectorologie. L'ADN nu tente en vain de confirmer tout l'espoir que l'on pouvait investir dans son utilisation vaccinale et le rêve ultime de la réparation in situ des gènes mutés n'est plus totalement irréaliste, mais ne parvient pas à
confirmer son potentiel par des résultats in vivo2.
En avril 2000, l’équipe française du Pr Alain Fischer à l’Hôpital Necker à Paris rapporte les premières observations convaincantes de rémissions complètes d'un déficit immunitaire
héréditaire (DICS-X1) sur 2 enfants inclus dans un essai clinique10. Ces résultats constituent la
première démonstration du potentiel thérapeutique de la TG. Il est à noter que sur les 20 patients enfants traités, 90% sont en rémission complète aujourd’hui avec un recul de plus de 10 ans11.
Malgré des signaux positifs, des coups durs ont provoqué des arrêts dans les développements et amené la communauté scientifique, et les autorités à plus de lucidité et de prudence. Parmi ces coups durs :
- 1999 : mort de Jesse Gelsinger (18 ans)12
. Cette patiente souffrait d’une déficience en ornithine transcarbamylase sous une forme non létale, et fut incluse dans un protocole de recherche de thérapie génique de cette maladie. Des déviations au protocole firent que Jesse Gelsinger, qui n’aurait pas dû être incluse, le fut malgré tout et reçut en plus une dose trop importante à cause d’un biais au protocole. Elle développa une forte réaction inflammatoire qui la plongea dans un coma, fatal après quelques jours. Une analyse post mortem a révélé un champignon respiratoire commun « Histoplasma capsulatum », et un virus HSV disséminé dans tout son corps. Aucun de ces microbes n’est normalement mortel, mais le système immunitaire de cette patiente pourrait avoir été affaibli par la
TG13 et contribué à la survenue de cette infection. L’adénovirus vecteur utilisé dans ce
protocole fut considéré comme une cause probable de cet événement indésirable.
- Fin 2002 : deux enfants atteint de SCID-X inclus dans l’étude de TG sur la DICS-X1 développent une prolifération incontrôlée de leurs Lymphocytes T, due probablement à
une mutagénèse insertionnelle dans un site « oncogène ». L’essai est arrêté jusqu’à ce
que l’identification des mécanismes responsables ait aboutie. L’un des patients meurt en Octobre 2003, l’autre récupère. La reprise de l’essai clinique est finalement autorisée après que le promoteur ait proposé un certain nombre de modifications au protocole comme la diminution du nombre de cellules administrées, l’âge des patients éligibles, la vérification d’antécédents familiaux hématologiques dans le but de réduire le risque d’oncogenèse insertionnelle. Faisant suite au développement d’un syndrome leucémique identique chez un autre enfant traité en janvier 2005, l’essai sera à nouveau arrêté de
Au cours des années qui suivront, 25% des sujets traités développeront ce type de prolifération cellulaire incontrôlée mais aucun autre décès ne surviendra. L’essai n’a toujours pas été redémarré. La FDA décidera de son côté de ne pas bloquer tous les essais sur la DICS-X1 mais de les encadrer avec une surveillance accrue. Ce choc fut le déclencheur de la décision d’élaboration de guidelines sur l’utilisation des vecteurs lentiviraux en thérapie génique et notamment de l’idée qu’il serait nécessaire de fixer un
nombre seuil de copies virales par cellule transduite15.
- 2007 : Décès de Jolee Morh (36 ans) une participante à un essai dans l’arthrite rhumatoïde :
Trois semaines après avoir reçu une seconde injection dans le genou d’un PTG expérimental afin de traiter son arthrite rhumatoïde, une jeune femme de 36 ans Jolee Mohr est retrouvée étendue inconsciente dans l’hôpital ou elle participait à son essai clinique à Chicago, respirant sous ventilateur alors que son foie était détruit. C’est sa famille qui décidât d’arrêter les systèmes de ventilation et de survie assistée 5 jours après qu’elle ait reçu sa deuxième injection. Encore plus grave que les dommages causés à son foie, une infection multi-résistante s’était déclenchée ayant causé une hémorragie interne, et un arrêt cardio-respiratoire. Le vecteur utilisé était un AAV pourtant reconnu comme bien toléré et pour ne pas déclencher de réponses immunitaires fortes chez l’animal, contrairement aux adénovirus, même s’il avait été déjà rapporté qu’une réponse humorale est déclenchée lors des premières administrations rendant difficiles les administrations répétées sans un traitement immunomodulateur auxiliaire.
Le décès de Jolee Morh a soulevé des questions éthiques13 :
pertinence de ce type d’approche émergente et présentant un niveau de risque
non négligeable pour des patients dont le handicap n’est pas évident. Pour le cas de cette patiente, son mari a rapporté que sa maladie était très peu invalidante, elle n’avait jamais eu le moindre souci pour jouer avec sa petite fille de 5 ans et dans son travail d’opératrice de saisie dont le salaire est basé sur la vitesse de saisie, elle était au sommet de l’échelle des salaires.
impartialité de l’investigateur quant à sa capacité à faire la part des choses entre
l’excitation scientifique suscitée par sa participation à la recherche de TG et l’intérêt pour le patient. Comme piste d’amélioration, le processus d’information des patients sur l’essai clinique conditionnant leur décision de participation pourrait dans le futur nécessiter l’intervention d’un médecin tiers qui ne connait pas le patient et le consentement du patient ne serait donc pas biaisé par la relation avec son médecin traitant.
Confusion pour le patient de la différence entre recherche et soin : le patient a
tendance à considérer que, puisqu’un médecin lui propose de participer à un essai, l’efficacité du traitement et sa sécurité sont garanties.
Nécessité au niveau des autorités de revoir au cas par cas chaque design plutôt
que sur une base de recommandations générales, et même parfois patient / patient.
D’après les données de mon examen probatoire et le travail objet de ce mémoire, entre 2007 et 2012 :
- 3 produits de Thérapie génique ont obtenu une autorisation de commercialisation respectivement la Gendicine et l’Oncorine en Chine dans le Cancer de la Tête et du cou et la Rexine-G aux Philippines dans le cancer Métastatique.
- Le nombre d’essais de Phase III en thérapie génique n’a atteint deux chiffres qu’à partir de 2008
- aucun PTG n’avait atteint le stade de l’AMM en Europe, aux Etats Unis ou au Japon. Le 19 juillet 2012, Alipogene tiparvovec (Glybera), un produit de thérapie génique développé par une société néerlandaise dans la déficience en lipoprotéine lipase, une maladie héréditaire très rare se traduisant par de graves crises de pancréatite est le premier produit de thérapie génique à obtenir un avis favorable de mise sur le marché de l’agence Européenne du Médicament. Le 25 octobre 2012 la Commission Européenne officialise cette autorisation16. 2012 devient donc une année historique pour la thérapie génique marquant la première autorisation de mise sur le marché en Europe d’un produit de Thérapie Génique.
Le stade des balbutiements a donc été dépassé et même si des coups durs sont survenus, ils ont eu au moins pour effet de recadrer les différents acteurs sur l’encadrement adéquat à donner à ces produits et essais novateurs. On peut ainsi faire le pari peu risqué que, dans les prochaines années, la TG, à côté de toutes les autres méthodes thérapeutiques qui bénéficieront elles aussi des rapides progrès en biologie, contibuera à l'amélioration du pronostic des maladies dont le traitement reste aujourd'hui insuffisant, ou viendra en remplacement de traitements efficaces mais très onéreux par des protéines recombinantes.
Ce projet d’ingénieur dont les bases ont été posées lors de mon mémoire probatoire fin 2009 a consisté à concevoir et tester un outil et sa méthodologie associée. Il s’agit d’un outil d’actualisation et de consolidation des développements en thérapie génique humaine permettant notamment de disposer d’une image à jour des PTG susceptibles d’atteindre le marché, à moyen et court terme. Ce besoin a été formulé initialement par la direction des Affaires Scientifiques de la filiale France de Genzyme fin 2008, afin de disposer d’une part d’une meilleure visibilité sur ces PTGs et d’autre part dans la perspective d’identifier d’éventuelles opportunités de développement externe. Après avoir été décrit dans mes objectifs début 2011, ce projet sera finalement abandonné par Genzyme, par une absence d’officialisation lors du rachat par Sanofi. Ayant besoin de temps pour appréhender des changements professionnels importants, je décide d’abord de mettre le projet en attente, puis, de le proposer à la société Astellas France lorsque j’intègre cette société en tant que Responsable Médical des Services et de la Qualité. Ce projet va recevoir un accueil favorable d’Astellas France qui devient un soutien. Aujourd’hui seule la société Astellas, moi-même et mes proches sont au fait des détails de ce projet. Une synthèse du travail réalisé sur ce projet, en tant qu’élève ingénieur, est fournie en annexe VI.
2. METHODOLOGIE EMPLOYEE
2.1. Périmètre
Le périmètre de ce travail a été élaboré selon les hypothèses et critères ci-dessous : - Choix du type de phases d’essais cliniques
De 1989 à avril 2013, on dénombre 1843 essais cliniques de Thérapie Génique (Graphique 1).
Graphique 1 : répartition des essais de TG par phase entre 1989 et avril 2013 selon la base Gene Therapy Clinical Trials Worldwide de John Wiley (consultée en avril 2013)
Les essais de Phase I et de Phase I/II représentent les premières administrations chez l’Homme de ces produits et permettent d’obtenir les premiers éléments de pharmacocinétique et de tolérance sans toutefois être prédictifs d’une réelle efficacité en situation réelle d’utilisation. Ils représentent 1451 essais cliniques de thérapie génique soit 79%. Compte tenu de la masse d’information à analyser et du très faible caractère informatif qui découlerait de son analyse en terme de projection de succès, ces essais de Phase I, de Phase I/II ou sur un seul patient n’ont pas été intégrés dans le périmètre d’analyse. Les essais de Phase IV ont également été écartés de l’analyse car ils concernent des produits déjà présents sur le marché et en conséquence en dehors du périmètre. Ce travail s’est donc focalisé sur les essais de Phase II, de Phase II/III et de Phase III qui représentent environ 20% (390) des essais de TG entre 1989 et avril 2013 mais qui représentent l’information la plus prédictive en termes de chances de succès des développements en cours pour les raisons principales suivantes :
Essais souvent menés en comparant les PTG avec des médicaments ou techniques
médicales de référence permettant d’apprécier l’efficacité et la tolérance des candidats
Nombre de patients et de sites suffisamment importants et différents pour limiter
- Types de PTG à l’étude :
Seuls les PTG répondant à la définition de l’ANSN ont été inclus dans le périmètre, c’est-à-dire les PTG transférés à l’homme quel que soit le vecteur utilisé ou la technologie de transfert mais surtout exprimés. Tous les essais sur des PTG non exprimés ou traduits comme les essais sur des PTG à base d’acides nucléiques antisens ont été mis de côté. De même tous les PTG visant à l’intégration et la production d’antisens in vivo ont été exclus. - Indications Thérapeutiques :
Aucune indication thérapeutique n’a été écartée à partir du moment où les critères précédents étaient respectés.
2.2. Matériel et Méthodes
2.2.1. Analyse de l’existant
Une vérification a d’abord été réalisée de l’existence ou pas de ce type d’outil soit en libre accès sur internet soit proposé par des sociétés. Ce type d’outil et de méthode ne semble pas être proposé à la date de ce mémoire.
Dans un deuxième temps, il a été vérifié l’existence de synthèses de qualité (exhaustives, documentées, justes et de portée équivalente) accessibles dans le domaine public traitant des produits de Thérapie Génique à potentiel d’accès au marché à moyen et court terme. Ce type de synthèse n’existe pas dans le domaine public. Les analyses publiques sont, de mon expérience, obsolètes ou non fiables et, ce qui est plus problématique, non remises à jour.
En revanche, des synthèses payantes et en général d’un coût assez élevé (aux alentours de 3 à 4000 $) existent sans que l’on puisse à l’avance bien déterminer leur contenu. Même si ces dernières présentent bien les tendances et succès, elles ne sont pas exhaustives et suffisamment précises, ni ne décrivent la méthodologie qui a permis de les réaliser si bien que leur utilisation stratégique est limitée.
2.2.2. Sources d’informations de référence
L’information sur les développements cliniques en cours et leurs résultats est une information considérée très confidentielle pour les entreprises réalisant ces développements, et plus particulièrement pour les PTG qui pourraient constituer un fort retour sur investissement pour peu que ces derniers atteignent le stade de la commercialisation. Si l’information sur l’existence des essais cliniques est « assez accessible » de par une généralisation des réglementations et recommandations imposant la publication d’éléments standardisés concernant ces essais sur des sites nationaux ou internationaux, on est loin de pouvoir en dire autant en ce qui concerne l’accessibilité de leurs résultats. La cause principale de cette difficulté à accéder aux résultats est que les réglementations dans le monde n’exigent pas toutes que les promoteurs d’essais cliniques rendent publics les résultats, et encore moins qu’ils les publient systématiquement dans des revues scientifiques.
Certains pays comme la France, intègrent dans leur réglementation l’obligation de rendre publique une synthèse des résultats mais c’est encore loin d’être une généralité au niveau mondial. En Europe par exemple, la généralisation de l’obligation de rendre publics les résultats des essais cliniques vient d’être entérinée le 27 mai 2014 avec la parution au JO européen du
règlement17 sur les essais cliniques mais cette exigence ne sera effective que lorsque toutes les
spécifications techniques prévues seront opérationnelles, notamment les portails web permettant de publier. Cette opérationnalité interviendra au plus tôt selon le texte en mai 2016. Il est à noter aussi que seuls des résumés des résultats des essais seraient publiés et qu’à ce stade il est impossible de pouvoir prédire la qualité des données qui seront mises à disposition. La FDA en 2007 dans son « Administration Amendments Act (FDAAA) » a demandé que les résultats des essais conduits aux Etats Unis des médicaments destinés à une AMM aux US soient rendus publics dans l’année suivant la fin de l’essai, que ces résultats soient par ailleurs publiés dans la littérature scientifique ou pas18.
Découlant de ce constat, une recherche des informations disponibles a abouti à la liste des sources suivantes par ordre de pertinence décroissante par rapport à la thématique :
- La base de données « Gene Therapy Clinical Trials Worldwide
(http://www.wiley.com/legacy/wileychi/genmed/clinical/) » mise en ligne par le journal de Médecine par les gènes (Journal of Gene Medecine) de l’éditeur Wiley,
- Les sites majeurs d’essais cliniques tenus à jour par les instances réglementaires au niveau international ou d’autres structures dont le plus connu, le plus ancien et le plus complet est le site américain « ClinicalTrials.gov » administré par le NIH (Institut National de la Santé). La liste des sites majeurs utilisés est fournie en annexe I.
- Les publications et revues scientifiques qui constituent une information de premier ordre lorsqu’elles existent pour un développement donné, ce qui est loin d’être le cas général comme évoqué précédemment,
- Les informations institutionnelles des entreprises notamment celles présentées sur leurs sites internet, les communiqués de presse de ces dernières, ou leurs présentations faites durant les manifestations scientifiques comme les congrès. Il est à noter que ces informations émanant des entreprises doivent être prises avec le recul qui s’impose et recoupées avec d’autres sources car elles sont par nature non indépendantes
- Les analyses financières de sociétés et des PTGs qu’elles développent constituent parfois la seule source d’information disponible. Ces sources correspondent aux informations et analyses utilisées par les investisseurs pour guider leurs choix d’investissements. Elles n’ont souvent pas la rigueur scientifique d’une publication mais souvent très informatives lorsque peu de données filtrent sur un développement donné et son état d’avancement. - Les analyses financières autres, puis enfin toute autre source traitant de la thématique,
lorsque les précédentes se sont révélées infructueuses.
Focus sur les modalités de mise à jour de la base “Gene Therapy Clinical Trials Worldwide” : Les données sont regroupées et régulièrement mises à jour par le journal de médecine génique (Journal of Gene Medecine) à partir des sources que sont les autorités compétentes officielles (RAC « recombinant DNA advisory committee », GTAC « Gene Therapy Advisory Committee etc..), la littérature publiée, les présentations faites lors de conférences et les informations fournies spontanément par les investigateurs et les promoteurs des essais eux-mêmes.
Ces derniers sont invités à transmettre tout nouvel essai de TG ou toute information concernant
un essai incomplet dans la base à l’adresse suivante « genemed@wiley.co.uk » pour prise en
compte. Les essais du monde entier sont pris en compte.
Comme l’information disponible sur certains essais est incomplète, l’approche standard adoptée par le journal est l’inclusion de tous les essais pour lesquels le pays et la maladie ciblée sont connus même si aucun autre élément n’est disponible. Même si l’approche consistant à ne pas toujours disposer du type de gène, de vecteur… n’est pas entièrement satisfaisante, elle présente l’avantage de disposer d’une vue d’ensemble fiable du nombre réel d’essais.
2.2.3. Procédure suivie
La base de données « Gene Therapy Clinical Trials Worldwide » a été utilisée pour : - lister tous les essais de thérapie génique conduits à ce jour.
- séparer les essais cliniques de thérapie génique selon leur phase II, II/III et III
Un fichier Microsoft Excel a été créé. Le choix d’Excel s’est fait sur sa simplicité d’utilisation et sa capacité d’adaptation au gré des besoins apparaissant au fur et à mesure du travail sur ce projet. L’idée était de concevoir un jeu de données le plus standard possible qui serait éventuellement transposable dans une base de données professionnelle lorsque les types de données à collecter seraient complètement identifiés.
Dans le fichier en question, des onglets différents ont été créés dans le but de différencier par la suite les essais :
- par phase de développement
- par statut des essais cliniques au sein de chaque phase de développement avec 3 statuts différents utilisés :
non encore initiés mais dont le démarrage est imminent (NYI) & en cours
terminés dont les résultats ont été un échec ou dont les résultats sont inconnus
terminés dont les résultats ont été un succès
La figure 2 illustre la répartition par onglets.
Figure 2 : onglets du fichier Microsoft Excel concernant les phases II
Une ligne a été créée dans le fichier par essai concerné. Pour chaque essai, tous les attributs de la base « Gene Therapy Clinical Trials Worldwide » ont été pris comme données de base d’identification.
Ces attributs sont les suivants :
- Identifiant unique de l’essai dans la base - Titre de l’essai
- Pays
- Type de pathologie (exemple : maladies cardiovasculaires) - Indication (exemple : maladie des artères coronaires) - Phase (II, II/III ou III)
- Date d’approbation ou d’initiation de l’essai - Statut (clos, en cours, pas encore démarré) - Date de clôture
- Gène transféré (exemple : FGF)
- Type de gène (exemple : facteur de croissance) - Vecteur (exemple : plasmide)
- Types de cellules véhiculant le vecteur en cas de TG ex vivo - Cellules cibles (exemple : cellules cardiaques)
- Type d’introduction du gène (in vivo, ex vivo)
- Voie d’administration (exemple : intra coronarienne) - Investigateur Principal
- Référence bibliographique
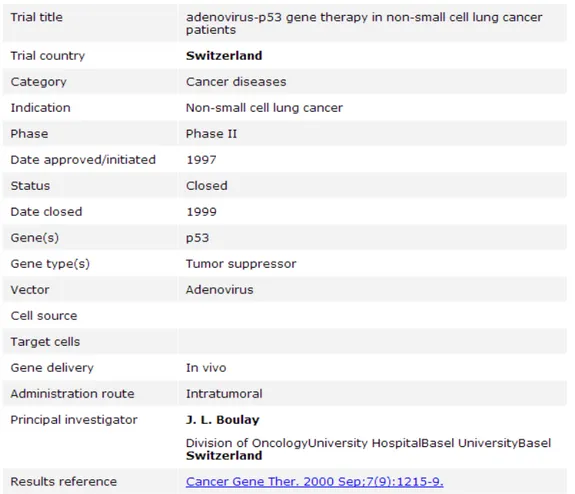
La figure 3 présente l’organisation d’une fiche essai source qui a été intégrée dans le fichier Excel.
Ces éléments d’identification standard sont nécessaires pour qualifier un essai mais pas suffisants pour décrire sa méthodologie. Par ailleurs, à moins que le nom ou un code du produit ne soit indiqué dans le titre, le produit n’est pas identifiable aisément avec ces attributs. En conséquence, pour pouvoir créer une image fidèle des produits et sociétés dont les développements cliniques sont avancés et les positionner les uns par rapport aux autres en fonction de leur état d’avancement, il a été nécessaire d’enrichir la base de données avec des éléments d’information supplémentaires concernant :
- le design de l’essai
- la société ou institution promoteur, dont le lien vers son site web le cas échéant, et le pays d’implantation
- Les références des produits concernés par l’essai
Après avoir initialisé le fichier avec l’ensemble des données contenues dans la base de données de référence « Gene Therapy Clinical Trials Worldwide », une partie conséquente du travail a donc consisté à collecter et valider les informations de design, d’identification des produits et des promoteurs.
Pour y parvenir :
- chaque essai issu de la base de données de référence a été recherché dans la base de données du NIH américain « ClinicalTrials.gov ». Lorsque l’essai a été retrouvé dans ClinicalTrials.gov sur la base d’une analyse de correspondances avec le titre, le gène, le vecteur et tout autre élément pertinent ; l’identifiant unique attribué dans ClinicalTrials.gov a alors été ajouté au fichier pour l’essai correspondant ainsi que le lien hypertexte vers la fiche descriptive de «ClinicalTrials.gov »
- lorsque l’essai n’a pas été identifié dans ClinicalTrials.gov, une recherche approfondie a été faite sur internet dans la littérature scientifique en utilisant le moteur de recherche Google scholar ou pubmed avec comme mots clés, tout élément pertinent de l’essai (exemples : le titre ou un code produit ou le nom de l’investigateur principal ou l’association de plusieurs d’entre eux).
- lorsque l’essai n’a pas été identifié dans la littérature scientifique à l’issue, alors les éléments ont été recherchés dans tout l’internet dans toutes les sources de référence citées au chapitre 2.2.2.
A chaque fois que le promoteur a été identifié à l’issue de ces recherches, les informations précédemment collectées ont été confirmées et consolidées en approfondissant les recherches autour des informations institutionnelles, financières et des communications scientifiques relayées par le promoteur ou par des tiers. Lorsque disponible, un lien a été ajouté dans le fichier vers le site internet du promoteur. Dans le cas de promoteurs institutionnels, il a été recherché à chaque fois si une société privée est associée au développement concerné ou si le développement est mené uniquement par une structure institutionnelle.
Par ailleurs, afin de limiter le risque de passer à côté d’un développement de Thérapie Génique en Phase II ou III, une alerte bibliographique a été mise en œuvre en parallèle avec le support du service documentation de Genzyme S.A.S. Les spécifications de cette alerte sont décrites en annexe II.
L’alerte a été mise en œuvre en mai 2009 jusqu’à la fin 2012. Les résultats de l’alerte m’ont été transmis chaque semaine. Ils étaient analysés systématiquement pour identifier dans les articles de nouveaux essais de thérapie génique. A partir de début 2013, l’alerte a été remplacée par un monitorage quotidien des newsletters pharmaceutiques reçues par le laboratoire Astellas Pharma.
Une autre étape clé de ce travail, et qui en fait son originalité, menée en parallèle de la première a consisté à enrichir les informations précédemment décrites (identification, design, produits, sociétés) avec les résultats des essais.
L’approche générale a été :
- de vérifier systématiquement la présence de résultats dans « ClinicalTrials.gov ». Le constat est que très peu de résultats d’essais sont finalement disponibles dans ClinicalTrials.gov pour les essais de Thérapie génique (moins de 1%).
- Une fois le promoteur identifié, il est possible de rechercher sur son site internet lorsque celui existe les publications scientifiques, présentations lors de congrès, les informations financières ou synthèses traitant de l’essai dont on recherche les résultats.
- Lorsqu’aucun résultat n’est identifié, la recherche de publications scientifiques via le site PubMed.gov, via Google Scholar ou sur internet au sens large permet d’obtenir des résultats. Si une publication payante contenant des résultats d’un essai est identifiée et qu’aucune autre donnée fiable publique et gratuite sur les résultats n’est disponible par ailleurs, la récupération de la publication a été demandée au service de documentation de Genzyme S.A.S (Cela n’a pas été nécessaire via Astellas) et la commande lancée ou enregistrée selon le prix du document. L’abstract lorsque disponible a été sauvegardé. - Enfin, si aucune donnée sur les résultats n’a été identifiée avec les méthodes précédentes,
des informations issues de communiqués de la presse financière ou d’autres médias financiers, les informations issues de forums internet spécialisés ont alors pu être considérées.
Les documents et publications scientifiques ont été indexés et reliés à chaque essai dans le fichier par des liens hypertexte, aussi bien pour les documents rendant compte du design, des produits, sociétés lorsque pertinents que pour les résultats intermédiaire ou finaux des essais. Une banque de documents a donc été constituée, connexe au fichier Excel. Cette banque, à la date de ce mémoire, compte plus de 500 documents. Les documents ont été classés par code unique d’essai dans le fichier suivi du signe « underscore » et d’un numéro d’ordre incrémentiel 1,2,3,4 etc... La figure 4 présente cette méthode d’indexation.
Figure 4 : indexation de la banque de documents en lien avec l’outil
Un statut a été attribué systématiquement afin de classer les essais terminés selon leurs résultats. Il s’agit de :
- Succès (lorsque les résultats sont statistiquement significatifs sur le critère principal de l’essai).
- Echecs (lorsque les résultats ne sont pas statistiquement significatifs) - Inconnus lorsqu’aucune information n’est disponible sur les résultats.
Pendant ces deux étapes précédentes, les erreurs et doublons identifiés dans la source de référence « Gene Therapy Clinical Trials Worldwide » de l’éditeur scientifique Wiley ont été supprimées au sein du fichier. A ce stade on peut qualifier le fichier de base de données dite « propre ».
La 3ème phase consiste à revoir chaque PTG utilisé dans chaque essai et à les qualifier
distinctement à l’aide des informations précédemment collectées selon leur(s) :
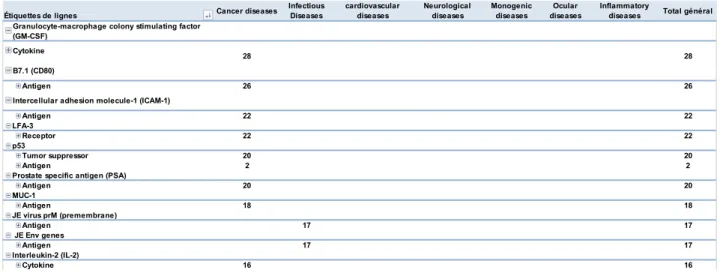
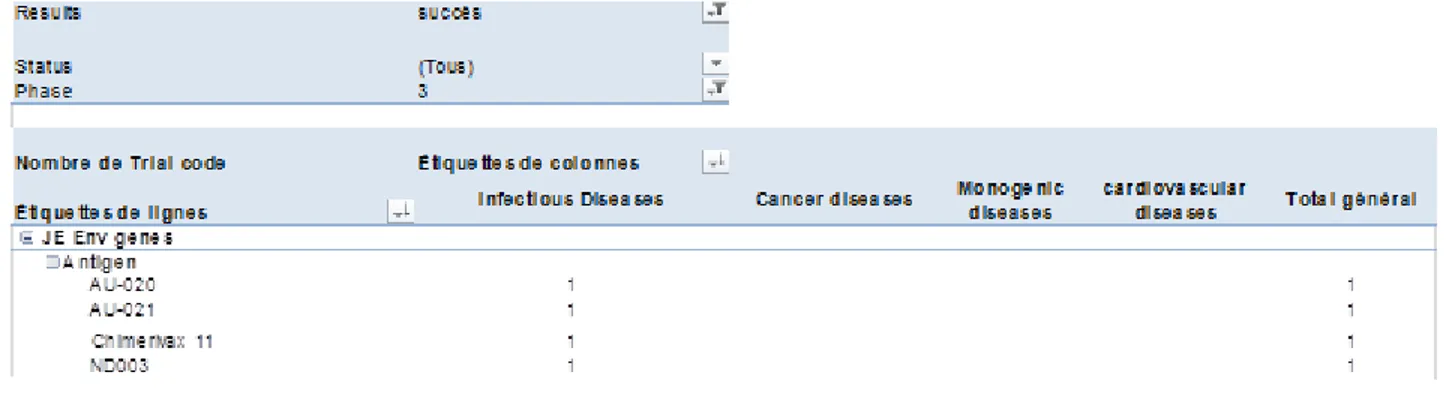
- Gène (s) : 1 PTG peut intégrer plusieurs gènes, chaque gène ayant son propre rôle thérapeutique au sein du produit
- Vecteur (s) : 1 PTG peut être composé de plusieurs vecteurs, chacun pouvant intégrer ses propres gènes. Certains vaccins thérapeutiques recombinants en sont un bon exemple mêlant plusieurs espèces virales vectorielles afin d’améliorer la réponse vaccinale recherchée.
- Véhicule (s) : Dans le cas des vecteurs complexes ou PTG préparés ex vivo (Figure 1), les vecteurs sont intégrés dans des véhicules par exemple des cellules autologues. Il peut également s’agir de plasmide encapsulé dans un véhicule lipidique (lipofection).
Une autre spécificité de ce travail est de permettre de classer les succès et les échecs par caractéristique(s) des PTG, tel que les vecteurs employés. La base de données « Gene Therapy Clinical Trials Wordlwide » est mise à jour tous les 3 à 6 mois par le journal de médecine génique, notamment pour y intégrer les nouveaux essais de TG identifiés. Les données nouvelles sont alors identifiées et intégrées dans la base de données consolidée, permettant ainsi :
- La mise à jour des informations des essais déjà répertoriés dans la base et notamment
leur statut,
La quatrième phase de la méthodologie consiste à isoler les produits dont les phases II & III pivotales sont un succès afin de constituer la cartographie des PTG ayant des chances d’atteindre le marché. Aujourd’hui, cette quatrième phase en est au stade du démarrage. Elle s’appuie sur les données collectées mais nécessite en plus une analyse non automatisée par produit. A l’issue de la quatrième phase, une fiche produit est créée pour chaque produit disposant d’un potentiel futur. Lorsque cette phase sera complètement terminée tous les produits concernés feront l’objet d’une fiche produit présentant successivement :
- Données générales pharmacotechniques du PTG, mécanisme d’action et indication visée - Grandes lignes de la clinique de la maladie et de son évolution, la thérapeutique standard,
le cas échéant les produits concurrents afin de positionner le produit par rapport à ces derniers
- Le plan de développement et les résultats obtenus
- Si applicable, le statut du dossier d’AMM et réglementaire du PTG vis-à-vis des autorités compétentes.
Les documents clés documentant ces statuts ont été recherchés et indexés le cas échéant. Pour ce faire, un dossier est créé par produit disposant d’une fiche produit et d’une banque de documents associés.
A la date du mémoire, seuls les produits ayant atteint le stade de la Phase III et les produits commercialisés de la période concernée ont fait l’objet de cette fiche. La fiche concernant le Glybera, premier produit de Thérapie Génique à avoir décroché une AMM Européenne est fournie en annexe III.
Les produits ayant atteint le stade de la mise sur le marché quels que soient le mode de distribution, le responsable de leur mise sur le marché, leur chiffre d’affaires et les pays concernés, sont par ailleurs considérés comme des succès. Ce critère de commercialisation d’un produit est un paramètre noté également dans la base au niveau de chaque essai associé au produit concerné indépendamment du succès de l’essai lui-même.
3. QUALITE DE LA BASE DE DONNEES
3.1. Eléments d’identification et de méthodologie des essais
Eléments méthodologiques :
La base de données réunit donc les essais de thérapie génique tels que définis précédemment de la Phase II à la Phase III et conduits dans le monde entre 1989 et avril 2013.
En avril 2013, 192 numéros d’identification CT.gov sont identifiés pour 283 études nettoyées de leurs doublons, soit 68% des essais répertoriés comportant une référence CT.gov et donc une description méthodologique approfondie. Si l’on étend l’analyse aux autres essais présentant un descriptif méthodologique même léger, on dénombre 216 essais sur 283 essais enregistrés soit 76 % des essais.
Promoteur et lien vers leur site web :
La base de données contient l’information relative aux promoteurs. Les promoteurs sont catégorisés selon qu’il s’agit de sociétés privées (pharmaceutiques ou de biotechnologies), d’établissements de santé ou de groupements d’établissements de santé (publics ou privés). Sur les 283 études que comporte la base, 270 études contiennent l’information du promoteur soit 95% des études. La bonne qualité de ce paramètre est clé afin de pouvoir disposer d’une image fiable des acteurs du secteur.
228 essais sur les 283 que contient la base, disposent du lien vers le site web du promoteur soit 81% des essais. Même si cet indicateur de qualité est biaisé par le fait que certains promoteurs ne disposent pas de site ou que certaines sociétés ont fait faillite et que le site n’est alors plus fonctionnel, cela reflète une bonne probabilité de disposer d’informations supplémentaires soit sur l’essai, soit sur le promoteur lorsque nécessaire.
Les produits concernés par les essais :
Les PTG investigués dans les essais cliniques de TG sont classés selon 3 niveaux :
- Un nom de produit commercial déposé ou non, attribué par la société ou l’institution qui le développe,
- et/ou un code produit.
- En l’absence de nom ou de code produit, un nom biomoléculaire est attribué. Le nom biomoléculaire est un nom générique qui n’est pas rattaché à l’entité qui le développe mais aux propriétés du PTG.
Il est composé de façon standard selon la convention suivante : gène puis vecteur puis véhicule. 194 essais comportent soit un nom de produit associé soit un code produit sur les 283 essais soit 69% des essais. Ce n’est pas pour autant d’ailleurs que l’on peut en conclure que le nom ou le code du produit n’a pas été trouvé dans 31% des essais. En effet, certains produits sont développés par des institutionnels qui n’attribuent pas forcément de noms ou codes lors de leurs développements propres. Ce chiffre est donc à mettre en perspective du pourcentage d’essais de TG menés par les institutionnels. Ce pourcentage s’élève à 70 essais sur les 283 que contient la base soit 25% des essais de TG, réduisant fortement la part des noms et codes produits réellement non disponibles dans la base.
3.2. Résultats des essais
Lorsque les essais sont en cours, ou pas encore démarrés, les résultats ne sont par essence pas disponibles. Un indicateur de disponibilité des résultats est donc mesuré en tenant compte de tous les essais clos, arrêtés prématurément ou suspendus pour lesquels les résultats peuvent être des succès, des échecs ou inconnus. Pour 86% des essais concernés, soit 186 essais sur 216, les résultats ont pu être déterminés.
Ces résultats démontrent une certaine performance du système dans la mesure où comme il a été évoqué dans les chapitres précédents, les réglementations demandant aux promoteurs d’essais cliniques de rendre publics les résultats des essais cliniques commencent juste à se mettre en place dans certaines régions du monde seulement et que dans tous les cas, aucune règle unifiée n’existe au niveau international. Par ailleurs, les promoteurs comme les journaux médicaux et scientifiques jusqu’alors sont peu enclins pour leur grande majorité à publier ou communiquer sur les résultats négatifs d’essais. Cela explique en partie la difficulté pour trouver de l’information pour les 14% de résultats non disponibles. Autre explication, certains résultats ne sont publiés que plusieurs années après la fin de l’essai et pourraient donc ne pas être encore publiés.
Information importante, une étude a montré en 2009 que moins de la moitié des essais contenus
dans clintrials.gov étaient au final publiés19. Ce résultat a été reconfirmé dans une étude récente
comparant la qualité de déclaration dans Clintrials.gov par rapport à la littérature scientifique20.
Ce chiffre de 50% théorique est bien inférieur aux 86% de résultats disponibles atteints grâce au recoupement de l’ensemble des sources disponibles.
4. RESULTATS
4.1. Résultats portant sur les essais cliniques
4.1.1. Distribution quantitative et qualitative par attributs (hors axes de recherches et indications)
4.1.1.1. Distribution par Phase et Statut
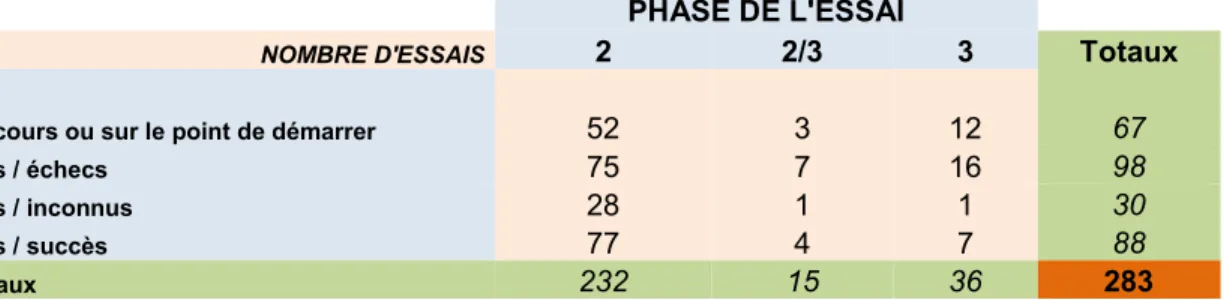
La distribution des essais par phase et statut est présentée dans le tableau 1 depuis les années 1990.
PHASE DE L'ESSAI
NOMBRE D'ESSAIS 2 2/3 3 Totaux
en cours ou sur le point de démarrer 52 3 12 67
clos / échecs 75 7 16 98
clos / inconnus 28 1 1 30
clos / succès 77 4 7 88
Totaux 232 15 36 283
Tableau 1 : Distribution des essais de TG en fonction de la phase et de leur statut depuis les années 1990 jusqu’à nos jours (données au 01 avril 2013)
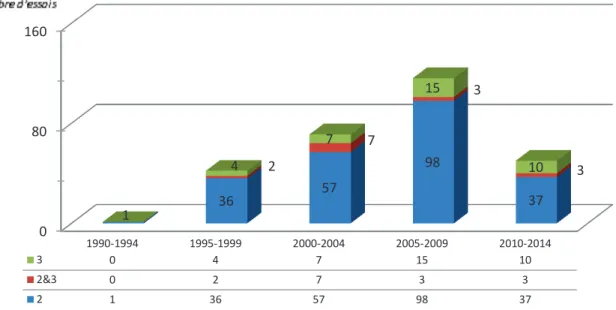
Le nombre d’essais au démarrage a augmenté régulièrement des années 1990 jusqu’à la première décennie 2000 puis semble marquer un repli à partir de 2010 (graphique 2).