INFLUENCE DU DIABETE DE TYPE II ET DE L'OBESITE A RISQUE SUR LA BIOTRANSFORMATION DES MÉDICAMENTS
Mémoire présenté
À la Faculté des études supérieures de l'Université Laval Dans le cadre du programme de maîtrise en pharmacie Pour l'obtention du grade de Maître es sciences (M.Se.)
FACULTE DE PHARMACIE UNIVERSITÉ LAVAL
QUÉBEC
2010
À mon grand-père Laurier Levac, un homme exceptionnel
[L'objectif de toute éducation devrait être de projeter chacun dans l'aventure d'une vie à découvrir, à orienter, à construire. ]
♦Albert Jacquard
[ Chaque fois que la science avance d'un pas, c'est qu'un imbécile la pousse, sans le faire exprès... ]
RÉSUMÉ LONG
Chez l'humain, les cytochromes P450 possèdent un rôle d'importance dans l'élimination des médicaments. Il existe plusieurs facteurs physiopathologiques qui sont en mesure d'affecter leur expression et leur activité. La présente étude a pour but de mieux comprendre le rôle de l'obésité à risque et du diabète de type II sur l'activité et l'expression des enzymes de biotransformation de la sous-famille CYP3A chez notre modèle, le cobaye. Ce modèle développé dans notre laboratoire est représentatif de l'état physiopathologique de l'obésité à risque et du diabète de type II. Pour ce faire les cobayes ont été soumis à des conditions nutritionnelles spécifiques pour une période de 190 jours.
Pour déterminer l'activité de cette sous-famille chez notre modèle, la dompéridone, substrat du CYP3A, a été utilisée lors d'incubations avec des microsomes hépatiques et intestinaux. Cette analyse a été effectuée par une technique HPLC spécifique pour détecter les metabolites majeurs formés par la sous-famille CYP3A, ainsi que pour détecter la présence de la molécule mère. Afin de déterminer l'expression protéique en plus de déterminer le dosage de l'ARN messager de la sous-famille CYP3A, des méthodes d'immunobuvardage de type western ainsi que des méthodes de PCR en temps réel ont été réalisées. Des diminutions significatives des 4 metabolites (Ml, M2, M3 et M4) de la dompéridone présents au niveau hépatique ainsi que des 2 metabolites présents au niveau intestinal (M3 et M4) ont été observées chez les cobayes soumis à une diète riche en sucrose ou fructose et en gras en comparaison avec nos animaux
sous diète contrôle. De plus, la quantification de l'ARN messager par méthode de PCR en temps réel a démontré une diminution significative de l'ARN messager de la protéine CYP3A au sein des deux groupes de cobaye avec diabète de type II et obésité à risque en comparaison avec nos témoins. Cependant aucune variation significative n'a été observée en ce qui concerne l'expression de la protéine CYP3A en immunobuvardage de type western et ce tant au niveau hépatique qu'intestinal. En conclusion, ces résultats suggèrent que l'activité enzymatique des CYP3A hépatiques et intestinaux serait diminuée en présence de l'état physiopathologique du diabète du type II et en présence d'une obésité à risque. Par conséquent, une biotransformation des substrats de la sous-famille CYP3A abaissée pounait par ailleurs entraîner au niveau clinique une augmentation des effets pharmacologiques, l'apparition d'effets indésirables, voire de la toxicité.
Chez l'humain, les cytochromes P450 possèdent un rôle d'importance dans l'élimination des médicaments. Il existe plusieurs facteurs physiopathologiques qui sont en mesure d'affecter leur expression et leur activité. La présente étude a pour but de mieux comprendre le rôle de l'obésité à risque et du diabète de type II sur l'activité et l'expression des enzymes de biotransformation de la sous-famille CYP3A chez notre modèle, le cobaye. Les résultats de la présente étude suggèrent que l'activité enzymatique des CYP3A hépatiques et intestinaux serait diminuée en présence de l'état physiopathologique du diabète du type II et en présence d'une obésité à risque. Par conséquent, une biotransformation des substrats de la sous-famille CYP3A abaissée pounait par ailleurs entraîner au niveau clinique une augmentation des effets pharmacologiques, l'apparition d'effets indésirables, voire de la toxicité.
REMERCIEMENT
Je tiens d'abord à remercier ma directrice ainsi que mon codirecteur de recherche, la Dre Chantale Simard et le Dr Benoît Drolet, pour m'avoir permis d'effectuer ma maîtrise dans un environnement extraordinaire au sein de leur équipe, pour leur disponibilité, leur humanisme, leur sens de l'humour sans oublier la confiance qu'ils m'ont apportée tout au long de ma formation. Je voudrais les remercier tout particulièrement pour leur support qu'ils m'ont apporté dans la réalisation de mes innombrables projets.
J'aimerais de plus, remercier Mme Sylvie Pilote ainsi que M. Dany Patoine pour leur aide en tant que professionnels de recherche. Merci de m'avoir soutenu moralement durant cette dernière année en plus de m'avoir permis d'acquérir des connaissances au sein du laboratoire. Je voudrais remercier Pascale Gagnon qui m'a apporté une grande aide dans la réalisation de mes travaux tout au long de la durée de ma maîtrise. Celle-ci a su m'appuyer et m'encourager dans les moments les plus difficiles. Je ne peux pas passer sous silence le support de mes grands-parents et de ma mère tout au long de mes études graduées. Je suis reconnaissant pour l'ensemble de l'aide financière que vous m'avez apportée sans oublier vos encouragements.
Finalement, ceux qui me connaissent bien savent que mes amis occupent une place importante dans ma vie. Vous m'avez permis tout au long de ma maîtrise de décrocher et vous m'avez permis de vivre des moments inoubliables.
Hélène, MP, MC, Éli, Caro, Guillaume, Geneviève, Kate et à tous ceux que je ne mentionne pas et que je considère mes amis, merci. Un merci tout particulier au Dr François Aube, mon ami et colocataire des dernières années pour l'ensemble de son aide apportée lors de la rédaction de ce mémoire.
Table des matières
RESUME LONG III RÉSUMÉ C O U R T V R E M E R C I E M E N T S VI LISTE DES TABLEAUX XI LISTE DES FIGURES XIII LISTE DES G R A P H I Q U E S XV LISTE DES ABRÉVIATIONS XVII 1. CHAPITRE 1 - INTRODUCTION : LES C Y T O C R H O M E S P450 1
1.1. INTRODUCTION-GÉNÉRALITÉ / 1.2. CARACTÉRISATION DES CYTOCHROMES. 5
1.2.1. Localisation et introduction générale 5 1.2.2. Mécanisme d'action général 6 1.2.3. Nomenclature et classification des cytochromes P450 7
1.2.4. La superfamille des cytochromes P450 10
1.2.4.1. La famille CYP1 10 1.2.4.1.1. La sous-famille CYPlA 10 1.2.4.2. La famille CYP2 .7 1.2.4.2.1. La sous-famille CYP2B . / 1.2.4.2.2. La sous-famille CYP2C 77 1.2.4.2.3. La sous-famille CYP2D 12 1.2.4.2.4. La sous-famille CYP2E 13 1.2.4.3. La famille CYP3 13 1.2.4.3.1. LeCYP3A4 14 1.2.4.3.1.1. La structure du CYP3A4 16 1.2.4.3.1.2. Polymorphisme et variance génétique 17
1.2.4.3.1.3. Implication et rôle endogène du CYP3A4 18
1.2.4.3.2. LeCYP3A5 18 1.2.4.3.3. LeCYP3A7 19 1.2.4.3.4. LeCYP3A43 19 1.2.4.3.5. Le CYP3A4 et la P-glycoprotéine (P-gp) 19
1.2.5. Régulation de la synthèse des cytochromes P450 21 1.2.5.1. Introduction sur l'induction et facteurs transcriptionnels 21
1.2.5.2. La superfamille des récepteurs nucléaires 24 1.2.5.2.1. Le récepteur nucléaire PXR 25 1.2.5.2.2. Le récepteur nucléaire CAR 26 1.2.5.2.3. Interaction entre PXR et CAR 26 2 . CHAPITRE 2 - INTRODUCTION : LE SYNDROME MÉTABOLIQUE 28
2.1. INTRODUCTION 28 2.1.1. Définitions et critères diagnostiques 28
2.1.1.1. L'obésité à risque 30 2.1.1.1.1. Influence de l'obésité à risque sur l'expression des CYP450 31
2.1.1.2. Le diabète 32 2.1.1.2.1. L'influence du diabète sur l'activité et l'induction des CYP450 35
2.1.1.2.1.1. Les études cliniques 35 2.1.1.2.1.2. Les études fondamentales 36
2.1.1.2.1.2.1. Les études animales 36 2.1.1.2.1.2.2. La théorie sur l'influence de l'insuline dans l'expression du
P450CYP3A4 38 3 . C H A P I T R E 3 - DESCRIPTION DE L ' É T U D E 41
3.1. Hypothèse générale 41 3.2. Objectif général 41
3.2.1. Objectifs spécifiques 41 4 . CHAPITRE 4 - LE COBAYE ET L'ÉLABORATION D'UN MODÈLE ANIMAL DE
SYNDROME MÉTABOLIQUE 42
4.1. INTRODUCTION 42 4.1.1. Le cobaye 42
4.1.1.1. Caractérisation de la courbe de croissance normale pour un cobaye mâle Hartley 43
4.1.1.2. Caractérisation de la phase 1 du métabolisme chez le cobaye 45 4.1.1.2.1. Séquence de nucleotides de l'ADNc pour les membres de la sous-famille
CYP3A 46 4.1.1.2.1.1. Alignement des différentes séquences de nucleotides entre l'humain et
le cobaye pour la sous-famille CYP3A 47 4.1.1.2.2. Séquence d'acides aminés de l'ADN codant pour les membres de la
sous-famille CYP3A 48 4.1.1.2.2.1. Alignement des différentes séquences d'acides aminés entre l'humain et
le cobaye pour la sous-famille CYP3A 48 4.2. STRA TÉG1ES ET MÉTHODOLOGIE UTILISÉES POUR L'ÉLABORA TION DU MODÈLE 49
4.2.1. Avant-Propos 49 4.2.2. La diète 50 4.2.3. Résultats 52 4.2.3.1. Évolution du poids 52 4.2.3.2. Glycémie provoquée 53 4.2.3.3. Dosage de l'insuline 54 4.2.3.3.1. Comptage de la radioactivité 56 4.2.3.3.2. Résultats 56 4.2.3.4. Bilan lipidique 56 4.2.3.4.1. Résultats 57 4.2.3.5. Coupes histologiques et aspect macroscopique (foie et reins) 61
4.2.4. Conclusion 64 5 . CHAPITRE 5 - EXTRACTION DE M I C R O S O M E S , QUANTIFICATION DES
CYP450 TOTAUX ET DOSAGE DES PROTÉINES TOTALES 66
5.1. Avant-propos 66 5.2. Méthodologie 66
5.2.1. Récolte des tissus 66 5.2.2. Extraction des microsomes hépatiques 66
5.2.3. Extraction des microsomes intestinaux 67
5.2.4. Dosage des protéines 68 5.2.5. Dosage des CYP450 totaux 7/
5.3. Conclusion 74 6. CHAPITRE 6 - ÉTUDE DE LA PROTÉINE CYP3A 75
6.1. ÉTUDE QUANTITATIVE DE L'ARN MESSAGER 75
6.1.1. Avant-propos 75 6.1.2. Méthodologie 75
6.1.2.1. Extraction de l'ARN 75 6.1.2.2. Dosage de l'ARN 76 6.1.2.3. Traitement de l'ARN 79 6.1.2.4. Synthèse de l'ADN complémentaire (ADNc) 79
6.1.2.5. PCR en temps réel 80 6.1.2.5.1. CYP450 3A 80
6.1.2.5.1.3.1. Expression de l'ARNm de la protéine P450 3 A hépatique 87
6.1.2.5.1.4. Conclusion 92 6.2. ETUDE DE L'EXPRESSION DE LA PROTEINE CYTOCHROME P450 3A 92
6.2.1. Avant-propos 92 6.2.2. Méthodologie 92
6.2.2.1. Immunobuvardage de type western 92 6.2.2.1.1. Préparation des échantillons 92
6.2.2.1.2. Résultats 95 6.2.2.1.3. Conclusion 96 6.3. ETUDE DE L'ACTIVITE 97
6.3.1. Avant-propos 97 6.3.2. Méthodologie 98
6.3.2.1. Incubations in vitro des microsomes hépatiques et intestinaux et analyse des
échantillons 98 6.3.2.2. Résultats 101
6.3.2.2.1. Résultats des analyses de l'activité du cytochrome P450 3A hépatique chez
nos différents groupes de cobayes 101 6.3.2.2.2. Résultats des analyses de l'activité du cytochrome P450 3A intestinale chez
nos différents groupes de cobayes 106
6.3.2.3. Conclusion 109 7 . CHAPITRE 7 - DISCUSSION 110 8. CHAPITRE 8 - CONCLUSION 118
Liste des Tableaux
Tableau 1 Classification générale des CYP450 impliqués dans la
biotransformation des médicaments chez l'humain 8 Tableau 2 Classification des CYP450 selon leurs substrats majeurs 9 Tableau 3 Enumeration des divers inducteurs du cytochrome P450 3A4 23 Tableau 4 Récepteurs nucléaires impliqués dans la modulation de
l'expression du cytochrome P450 3A4 24 Tableau 5 Listes des divers critères énoncés par Y «ATPIII» et l'Organisation
mondiale de la santé (OMS) pour émettre un diagnostic de
syndrome métabolique 29 Tableau 6 Ensemble des articles obtenus lors d'une revue de littérature
portant sur l'influence du diabète sur l'expression du P450
CYP3A4 37 Tableau 7 Données représentatives moyennes du poids en fonction du temps
pour un cobaye mâle Hartley 44 Tableau 8 Cytochromes P450 du cobaye répertoriés par le site
www.uniprot.org 45 Tableau 9 Pourcentage d'homologie entre les différentes séquences de
nucleotides entre l'humain et le cobaye pour la sous-famille
CYP3A 47 Tableau 10 Pourcentage d'homologie entre les différentes séquences d'acides
aminés entre l'humain et le cobaye pour la sous-famille CYP3A 48 Tableau 11 Information nutritionnelle des trois diètes utilisées lors du
protocole animal 51 Tableau 12 Constitution des différents éléments de dosage pour l'analyse RIA
de l'insulinémie chez le cobaye 55 Tableau 13 Trousses de réactifs utilisées dans les différentes analyses
effectuées par le laboratoire de biochimie de 1TUCPQ 57 Tableau 14 Résultats des divers paramètres biologiques obtenus après 190
jours de diète spécifiques chez nos trois groupes de cobayes à
l'étude 60 Tableau 15 Enumeration des divers critères supportant l'hypothèse d'un
Tableau 16 Résultats du dosage des protéines pour nos tissus hépatiques et
intestinaux pour l'ensemble de nos animaux à l'étude 70 Tableau 17 Séquences d'oligonucléotides utilisées dans les essais de PCR en
temps réel 81 Tableau 18 Moyenne et données en duplicata des différents «CT» corrigés et
obtenus à la suite de l'amplification de l'ADNc de la protéine
cytochrome P450 3A 88 Tableau 19 Moyenne et données en duplicata des différents «CT» corrigés et
obtenus à la suite de l'amplification de l'ADNc de la protéine
calnexine 89 Tableau 20 Divers paramètres calculés de ACT et AACT à partir des valeurs de
«CT» conigées et le ratio d'expression finale de l'ARNm de la sous-famille cytochrome P450 3A pour nos trois différents groupes
de cobayes 90 Tableau 21 Ratios d'expression finaux moyens de l'ARNm de la sous-famille
cytochrome P450 3A pour nos trois différents groupes de cobayes 91 Tableau 22 Protocole d'utilisation des anticorps en immunobuvardage de type
western 94 Tableau 23 Activité des CYP3A hépatiques - Ratio de la hauteur des
metabolites de dompéridone formés sur la hauteur du standard
interne 104 Tableau 24 Activité des CYP3A intestinaux - Ratio de la hauteur des
metabolites de dompéridone formés sur la hauteur du standard
interne 107 Tableau 25 Autres facteurs et tests biochimiques pouvant nous permettre de
Liste des figures
Figure 1 Principales enzymes impliquées dans la biotransformation de phase I
et de phase II des médicaments 2 Figure 2 Schéma résumé du métabolisme des xénobiotiques 4
Figure 3 Représentation schématique de la structure d'un cytochrome P450 5 Figure 4 Schéma de la réaction générale sous forme de cycle catalytique 7 Figure 5 Structure tridimensionnelle du CYP3A4 et de son site actif 16 Figure 6 Facteurs pouvant influencer l'activité des récepteurs nucléaires 22 Figure 7 Autres interactions possibles pouvant influencer l'activité des
récepteurs nucléaires CAR et PXR 27 Figure 8 Évolution de l'état pathologique du diabète de type II 33
Figure 9 Estimation du nombre d'adultes atteints de diabète par groupe d'âge
pour les années 2000 et 2030 dans le monde 34 Figure 10 Représentation schématique du rôle de l'insuline dans la régulation du
métabolisme du glucose et des lipides via les facteurs de transcription Foxol (Forkhead box protein Ol) and Foxa2 (Forkhead box protein
02) 38 Figure 11 Illustration de l'implication de la voie de signalisation de l'insuline
dans la régulation de l'expression du P450 CYP3A4 40 Figure 12 Différences macroscopiques du foie et du rein chez nos différents
groupes de cobayes 61 Figure 13 Image microscopique démontrant la présence d'une légère stéatose
hépatique chez nos cobayes avec une diète riche en gras et en sucres 62 Figure 14 Image microscopique démontrant la présence de calcification rénale
chez nos cobayes avec une diète riche en gras et en sucre 63 Figure 15 Spectre entre 400 et 500 nm à la suite de la saturation d'un échantillon
de microsomes du cobaye numéro 3 en monoxyde de carbone (CO) et
de l'ajout du dithionite de sodium (Na2S2Û4) 72 Figure 16 Imagerie obtenue suite à la migration de 2 \ig d'ARN de foie de
Figure 17 Photo obtenue suite à la migration de 2 u.g d'ADNc sur un gel
d'agarose 1% pour l'ensemble de nos 22 cobayes à l'étude 83 Figure 18 Représentation sur film d'un immunobuvardage de type Western de la
protéine cytochrome P450 3A et du standard interne chez nos trois
différents groupes de cobayes 95 Figure 19 Principaux metabolites de la dompéridone suite à une
biotransformation par le cytochrome P450 3A4 98 Figure 20 Chromatogramme obtenu suite à l'analyse HPLC d'une incubation de
microsomes hépatiques en présence de dompéridone pour un cobaye
témoin 103 Figure 21 Chromatogramme obtenu suite à l'analyse HPLC d'une incubation de
microsomes intestinaux en présence de dompéridone pour un cobaye
Liste des Graphiques
Graphique 1 Poids en fonction du temps chez les divers groupes de cobayes (sucrose, fructose et témoin) à partir de leurs arrivées au
laboratoire à 3 semaines d'âge 53 Graphique 2 Glycémie en fonction du temps lors d'un test d'hyperglycémie
provoquée oral (HGPO) chez nos animaux 54 Graphique 3 Cholestérol total en fonction du temps pour nos trois groupes
de cobayes à l'étude 58 Graphique 4 Concentration des triglycérides en fonction du temps pour nos
trois groupes de cobayes à l'étude 58 Graphique 5 Concentration en urée en fonction du temps pour nos trois
groupes de cobayes à l'étude 59 Graphique 6 Résultats moyens obtenus, pour nos trois différents groupes de
cobayes (témoin, HFSD et HFFD) lors de la quantification des
CYP450 totaux 73 Graphique 7 Ratios finaux moyens d'expression de l'ARNm de la
sous-famille cytochrome P450 3A pour nos trois différents groupes
de cobayes 91 Graphique 8 Résultats obtenus, pour nos trois différents groupes de cobayes
(témoin, HFSD et HFFD), lors de la lecture du film ci-haut en
densitométrie 96 Graphique 9 Ratio hauteur du métabolite/hauteur propranolol pour les 4
metabolites obtenus lors de l'analyse en HPLC des incubations de microsomes hépatiques de cobaye pour les 3 diètes :
témoin, HFSD et HFFD 105 Graphique 10 Ratio hauteur du métabolite/nauteur propranolol pour les 2
metabolites obtenus lors de l'analyse en HPLC des incubations de microsomes intestinaux de cobaye pour les 3 diètes :
Liste des annexes
Annexe 1 Caractérisation du cytochrome P450 CYP2B chez le cobaye 128 Annexe 2 Caractérisation du cytochrome P450 CYP2C chez le cobaye 130 Annexe 3 Caractérisation du cytochrome P450 CYP2E1 chez le cobaye 133 Annexe 4 Séquence de nucleotides pour les membres de la sous-famille
CYP3A chez le cobaye 136 Annexe 5 Séquence d'acides aminés de l'ADN codant pour les membres
Liste des abréviations
1. ADNc : Acide désoxyribonucléique complémentaire 2. CO : Monoxyde de carbone
3. CYP450 : Cytochrome P450
4. CYP3A : Cytochromes P450 de la sous-famille 3A 5. CRP : C-reactive protein
6. DMSO : diméthylsulfoxyde
7. EDTA : Ethylene diamine tetra acetic acid 8. ER : Estrogen receptor
9. FAD : flavine adenine dinucléotide 10. FMN : Flavine mononucleotide 11. FOXOl : factor Forkhead Box 01 12. FOX02 : factor Forkhead Box 02 13. FXR : farnesoid X receptor 14. GR : glucocorticoid receptor 15. HDL : high-density lipoprotein 16. HIF : Hypoxia-inducible factors 17. HNF-4 : Hepatocyte Nuclear Factor 4
18. HPLC : High performance liquid chromatography, aussi appelé high pressure liquid chromotography
19. HFSD : High fat high sucrose diet 20. HFFD: High fat high fructose diet 21. IMC : Indice de masse corporel
22. IUCPQ : Institut universitaire de cardiologie et de pneumologie de Québec 23. LXR : Liver X receptors
24. MCV : Maladie cardiovasculaire
25. NADP+ : Nicotinamide-adénosine-dinucléotide-phosphate
26. NADPH : Nicotinamide-adénosine-dinucléotide-phosphate (réduit) 27. PB : Classe pharmacologie des phénobarbituriques
28. PBS : Phosphate buffered saline
29. PPAR : peroxisome proliferator-activated receptor 30. RIA : Radio immuno assay
31. SHP : Short heterodimer partner 32. TG : Triglycéride
33. TR : Thyroid hormone receptor
34. TSOD : Tsumura, Suzuki, obese, diabètes mice 35. VIH : Virus d'immunodéficience humaine 36. VDR : Vitamin D Nuclear Receptor
1.1. INTRODUCTION-GÉNÉRALITÉ
Les êtres humains sont tous les jours exposés à plusieurs agents étrangers que l'on nomme xénobiotiques. Ces substances sont absorbées à partir de la peau, des poumons ou la plupart du temps ingérées de façon non-intentionnelle à partir de la nourriture ou encore délibérément comme médicaments à des fins thérapeutiques ou encore comme drogue d'abus. Certains sont inoffensifs, mais certains pounont entraîner une réponse biologique.1 Au cours de l'évolution, les êtres vivants ont développé des systèmes
limitant l'accumulation de ces composés afin de protéger l'intégrité de l'organisme. Cette protection contre les xénobiotiques est rendue possible par la présence au sein de l'organisme de transporteurs membranaires permettant une sortie rapide des molécules indésirables et de systèmes enzymatiques de conversion chimique permettant de rendre les molécules plus hydrophiles afin de faciliter leur élimination dans les fluides corporels.2
Le foie est l'organe majeur responsable du métabolisme des xénobiotiques.2' 3
Toutefois, d'autres organes comme le tractus gastro-intestinal, les poumons, et les reins en autres, peuvent également participer à leur biotransformation.4 La nature chimique de
ces composés étrangers peut être très variable et imprévisible. C'est pour cette raison que l'organisme possède de nombreux enzymes, isoenzymes et transporteurs pour être capable de métaboliser l'ensemble des xénobiotiques potentiels.5 Ces enzymes et
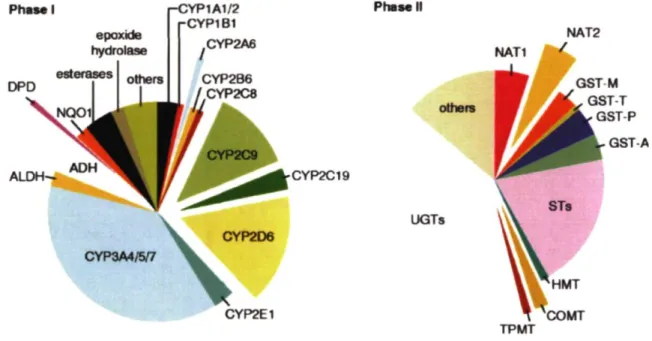
(Figure 1) et la phase III incluant les transporteurs.2
Figure 1. Principales enzymes impliquées dans la biotransformation déphasé I et de phase II des médicaments. Adapté d'Evans et Retting. Science. 1999.6
PhSMl DPD epoxide hydrolase I-CYP1A1/2 rCYP1B1 Phase II NAT2 L, GST A ALDH^ CYP2C19 UGTs CYP3A4/5/7 CYP2E1 TPMT
Les réactions de phase I convertissent habituellement le produit mère en un metabolite plus polaire soit par oxydation (désalkylation, désamination, déshydrogénation, hydrogénation), par réduction ou par hydrolyse.7 Si le metabolite
formé a atteint un degré d'hydrosolubilité suffisant, l'élimination pouna donc se faire rapidement. Cependant, dans la situation contraire, le metabolite pouna subir des réactions subséquentes et poursuivre son processus de biotransformation par la phase II. Lors de cette phase, des substance endogènes comme l'acide glucuronique (glucuronidation), l'acide sulfurique (sulfatation), l'acide acétique (acetylation), le gluthation ou encore certains acides aminés se combinent avec les nouveaux groupements
est à noter que la phase I n'est pas obligatoire et que certaines molécules peuvent subir immédiatement la phase II. En bref, les réactions de phase I peuvent être considérées comme des réactions de modification ou de conversion de groupements fonctionnels, tandis que les réactions de phase II constituent des réactions de conjugaison de groupements fonctionnels.
Tel que mentionné ci-haut, la phase III du métabolisme regroupe les transporteurs qui facilitent l'élimination des xénobiotiques. La superfamille des ATP-binding cassette (ABC) est une classe de transporteurs qui permet le transport de plusieurs éléments biologiques ainsi que de composés pharmacoactifs à travers la membrane. Ceci est possible suite à l'hydrolyse de l'ATP, ce qui permet une translocation du composé contre un gradient de concentration. Une des protéines de transport les plus importantes dans le domaine de la pharmacocinétique est connue sous le nom de P-glycoprotéine (P-gp) (ABCB1).2 Celle-ci fut décrite pour la première fois dans les années 70 par V. Ling.10' n
Son poids moléculaire est de 170 kDa; elle est codée chez l'être humain par le gène 17 •
MDR1. La P-gp agit comme une pompe transmembranaire qui provoque un efflux d'agents toxiques hors de la cellule. Sa présence au niveau intestinal et au niveau hépatique permet un efflux des composées dans la lumière intestinale et dans la bile respectivement.13
phases de biotransformation et d'élimination d'un xénobiotique. Cette molécule de nature hydrophobe (SH) pénètre à l'intérieur de la cellule ou elle subira une hydroxylation (Phase I). Le metabolite généré (SOH) sera par la suite transformé en un deuxième metabolite par la conjugaison d'un nouveau groupement X (SOX) (Phase II), pour finalement être éliminé par un transporteur membranaire (Phase III).
Figure 2. Schéma résumé du métabolisme des xénobiotiques. Adapté de Beaune. Thérapie. 1993.5
Phase III Elimination
^
x
__„
Les cytochromes P450 (CYP450) sont des protéines membranaires principalement situées au niveau du reticulum endoplasmique lisse. Ces enzymes se retrouvent tant chez les mammifères et les plantes que chez les invertébrés14. Les CYP450 ont été rapportés
pour la première fois par Omura et Sato en 1964.15 Ils représentent les enzymes les plus
importantes dans la biotransformation des médicaments. Us sont des hémoprotéines d'environ 50 kDa de masse moléculaire, possédant une molécule d'hème attachée à l'apoprotéine par une liaison nommée thiolate qui s'établit entre le fer et le soufre d'un résidu cysteine situé proche de la portion C-terminale de la protéine (figure 3).
Figure 3. Représentation schématique de la structure d'un cytochrome P450. Adapté de Beaune. Thérapie. 1993.5
Liaison thiolate
M = Hème
P.M. = 50 000
Site actif hydrophobe
La partie N-terminale de la protéine est constituée d'acides aminés hydrophobes permettant à celle-ci de se fixer au niveau de la membrane. L'ensemble des CYP450 constituent une superfamille de 57 gènes possédant tous le même groupe actif, l'hème, mais différents par la structure de leurs apoprotéines. Le nom de cytochrome P450
lorsqu'ils se retrouvent sous une forme réduite et lorsque la structure de l'hème est associée avec une molécule de monoxyde de carbone.
1.2.2 MÉCANISME D'ACTION GÉNÉRAL
Ces enzymes sont aussi appelées mono-oxygénases, car elles incorporent un atome d'oxygène à partir de l'oxygène moléculaire. Pour ce faire, celles-ci doivent être associées avec une flavoprotéine appelée NADH-cytochrome P450 reductase qui permet le transfert des électrons du NADPH aux cytochromes P450. La réaction générale d'oxydation d'un substrat (R) catalysée par le CYP450 peut être représentée comme suit :
NADPH + H+ + 02 + R ► NADP+ + H20 + RO
De façon plus complexe, la réaction générale peut être représentée schématiquement sous la forme d'un cycle catalytique (Figure 4). À l'étape 1, le substrat se lie à l'enzyme au niveau de la région de l'hème en libérant ainsi une molécule d'eau. A l'étape 2, l'électron provenant du NADPH est rendu disponible grâce à l'intervention des flavoprotéines NADH-P450 reductase, en particulier FAD et FMN. L'étape 3 se caractérise par la liaison de FO2 au CYP450 maintenant sous l'état de Fe2+ dû au gain de
l'électron à l'étape 2. Suivant la liaison avec l'oxygène, le cytochrome b5, non présent lors de la première réduction trop thermodynamiquement difficile, permet une deuxième réduction en fournissant un électron supplémentaire (étape 4). L'introduction de deux atomes d'hydrogène aux étapes 5 et 6 aura comme effet de cliver le lien 0 - 0 et de libérer
forme un complexe avec le substrat. À l'étape 8, le produit de réaction est généré par la dissociation du substrat de l'enzyme par l'introduction d'une molécule d'eau.16"18
Figure 4. Schéma de la réaction générale sous forme de cycle catalytique. Adapté de Werk. Genomebiology. 2000.
H;° « m w w <•
X
*______ J ^ r r^TC °%"
s s s
y x y s
Cy» . - . ^ ^ A Cy* . Cy»•■.... /
^ é W :' Cy» .
• f c ^
B
'Je-_ ^ - r - ^ 7 *V- ^W ^ — ^ 7
zs x s ici x s n»r /s IAT
Cy».... <£*-«-. c» » . Cy»...
7.2.5 NOMENCLATURE ET CLASSIFICATION DES CYTOCHROMES P4 5 0
Les cytochromes P450 sont identifiés à partir d'une nomenclature précise selon une recommandation universelle. Pour commencer, le gène chez l'humain est représenté symboliquement en italique par les lettres « CYP », alors que l'enzyme de son côté est décrite par le symbole « CYP ». Par la suite, dans les deux cas, ce symbole est suivi d'un chiffre arabe représentatif de la famille suivi d'une lettre majuscule désignant la sous-famille et d'un second chiffre arabe représentant le gène individuel à l'intérieur de cette
sous-famille. L'ensemble des protéines d'une même famille possède entre elles une similitude d'au moins 40% au niveau de leur séquence protéique. De plus, les cytochromes P450 d'une même sous-famille doivent démontrer dans leur séquence protéique une similitude supérieure à 55% (tableau 1).
Tableau 1. Classification générale des CYP450 impliqués dans la biotransformation des médicaments chez l'humain. D'après Beaune 1993.5
F. mille > 40°. similitude s F. s 1 2 3 4 A B A B C D E F A A B F 1,2 1 6,7, 13 1,2,6 8,9,10. 17,18, 19 6 1 1 3,4,5,7 9.11 1 2,3
Dans le cas d'une découverte d'une nouvelle séquence, pour que celle-ci soit reconnue comme un nouveau gène individuel, la séquence de ce dernier doit être différente d'au moins 3% par rapport aux autres gènes existant dans la sous-famille.18
Outre la nomenclature, il existe plusieurs méthodes établies pour classifier les CYP450. Il est possible de classifier ceux-ci selon leurs substrats majeurs endogènes soient : les sterols, les xénobiotiques, les acides gras, les eicosanoïdes et les vitamines (tableau 2). Les cytochromes dont nous ne connaissons pas les substrats sont classifies dans une classe à part 19
Sterols Xénobiotiques Acides gras
Eicosanoïdes Vitamines non identifiés
1B1 1A1 2J2 4F2 2R1 2A7 7A1 1A2 4A11 4F3 24A1 2S1 7B1 2A6 4B1 4F8 26A1 2U1 8B1 2A13 4F12 5A1 26B1 2W1 11A1 2B6 8A1 26C1 3A43 11B1 2C8 27B1 4A22
11B2 2C9 4F11
17A1 2C18 4F22
19A1 2C19 4V2
21A2 2D6 4Z1
27A1 2E1 20A1
39A1 2F1 27C1
46A1 3A4 51A1 3A5 3A7
Notamment, les CYP450 peuvent aussi être divisés en quatre classes indépendantes basées sur le moyen par lequel l'électron provenant du NADPH est délivré au niveau du site catalytique de l'enzyme. Dans la classe I, la protéine requiert la présence simultanée d'une flavoprotéine NADPH-P450 reductase, soit la FAD et la présence d'un atome de fer-soufre réduit. Dans la classe 2, la protéine nécessite seulement la présence d'une flavoprotéine NADPH-P450 reductase, soit la FAD ou la FMN pour pouvoir transférer un électron. Dans la classe III, aucun donneur d'électron n'est nécessaire au bon fonctionnement de la protéine. Finalement, pour ce qui est de la dernière classe, la classe IV, les électrons sont directement fournis par le NADPH.16
1.2.4 LA SUPERFAMILLE DES CYP450 1.2.4.1 LA FAMILLE CYP1
La famille CYP1 est constituée de la sous-famille CYP1A et de la sous-famille CYP1B.
1.2.4.1.1 LA SOUS-FAMILLE CYP1A
Cette sous-famille comporte 2 enzymes spécifiques partageant une homologie de 70% au niveau de leur séquence d'acides aminés.20 Les gènes de ces enzymes, le
CYP1A1 et le CYP1A2, sont situés sur le chromosome 15q22-q24.21'22 L'expression du
CYP1A1 est principalement extra-hépatique et se concentre surtout, suivant une induction, dans des tissus comme les poumons, les seins, l'utérus, le colon et le placenta.5
Celui-ci participe très peu à la biotransformation des médicaments, ce qui en fait un cytochrome peu important au niveau de la pharmacocinétique clinique. Son rôle principal est de métaboliser les polluants environnementaux tels que le benzo(a)pyrène, un hydrocarbure aromatique que l'on retrouve dans le tabac.
Quant au CYP1A2, il est essentiellement exprimé au niveau du foie et représente 13% de l'ensemble des cytochromes hépatiques. Cette protéine de 515 acides aminés
71
avoisine les 58 kDa de poids moléculaire. De plus, il est impliqué dans la biotransformation de plusieurs médicaments, stéroïdes et de certains polluants environnementaux. La furafylline est un inhibiteur spécifique du CYP1A2.23 Cet
inhibiteur permet la discrimination avec le CYP 1 Al lors d'études pharmacocinétiques. En outre, les molécules ayant une structure connexe à l'agent anti-arythmique lidocaïne et plusieurs médicaments de type quinolol inhibent l'activité du CYP1A2 in vivo et in
vitro.24 Quelques évidences épidémiologiques confirment qu'une activité enzymatique
augmentée au niveau du CYP1A2 corèle fortement avec un risque augmenté de cancer du colon.25
1.2.4.2 LA FAMILLE CYP2
La famille CYP2 est reconnue comme étant la plus grande des familles. Celle-ci possède aussi une des plus grandes diversités en termes d'enzymes. Elle comprend 6 sous-familles CYP2A, CYP2B, CYP2C, CYP2D, CYP2E et CYP2F.
1.2.4.2.1 LA SOUS-FAMILLE CYP2B
La sous-famille CYP2B se retrouve en faible quantité dans le cerveau, les poumons, les intestins, les reins et le foie particulièrement en absence d'induction. Certains membres de cette sous-famille possèdent la caractéristique d'être induits par des xénobiotiques dont la classe des phénobarbituriques (PB). Les gènes de ce groupe sont situés sur le chromosome humain 19ql2-13.2.21 Le cytochrome CYP2B6 que l'on
retrouve chez l'humain serait responsable de la bioactivation de certains anticancéreux comme la cyclophosphamide et de la biotransformation du bupropion, agent utilisé pour l'anêt tabagique.26 De plus, nous retrouvons également chez l'humain dans cette même
sous-famille le cytochrome 2B1/2 se localisant au niveau de l'encéphale. Ceux-ci sont induits par la nicotine et permettent la biotransformation de la morphine. '
1.2.4.2.2 LA SOUS-FAMILLE CYP2C
Étudiée depuis plusieurs années, cette sous-famille est la seconde en importance après la sous-famille CYP3A. Elle est composée de 4 gènes : CYP2C8, 2C9, 2C18 et
2C19, qui se partagent environ 80% de leur séquence d'acides aminés. Parmi eux, 1'isoenzyme CYP2C9 est la plus abondante, suivie du CYP2C8 et du CYP2C19. Représentant 18 % des enzymes totales au foie, cette sous-famille est également présente au niveau de l'intestin. En plus de métaboliser plusieurs composés endogènes, ce système metabolise approximativement 25 % des médicaments se retrouvant sur le marché, ce qui en fait un groupe d'enzymes d'importance clinique. Les polymorphismes génétiques présents au sein de cette sous-famille exercent un impact clinique non négligeable. Ces polymorphismes, affectant surtout les enzymes CYP2C9 et CYP2C19, se traduisent par une variation de la vitesse de biotransformation interindividuelle. 29~31
1.2.4.2.3 LA SOUS-FAMILLE CYP2D
Chez l'humain, le CYP2D6 est le seul gène actif de la sous-famille CYP2D qui comprend deux autres membres soit les pseudogènes CYP2D7 et CYP2D8. L'expression du CYP2D6 se retrouve majoritairement au niveau du foie, bien qu'on le retrouve au niveau de l'intestin et des reins. Tout comme certains membres de la sous-famille CYP2C, il existe certains polymorphismes génétiques affectant l'activité de l'enzyme CYP2D6, ce qui en a fait l'un des cytochromes les mieux caractérisés. De ces changements génétiques découlent les phenotypes de métabolisateurs rapides ou lents. Les métabolisateurs rapides représentent somme toute environ 90-95% de la population caucasienne. Ceux-ci possèdent au niveau de leur bagage au moins un allele fonctionnel du CYP2D6. De ce groupe, environ 5% des individus auront de multiples copies du gène ce qui en fera des métabolisateurs ultrarapides. À l'inverse, l'altération complète de l'expression du gène entraînera le phénotype des métabolisateurs lents. Cette variabilité
dans l'activité de l'enzyme affecte grandement la biotransformation des composés substrats du CYP2D6. Ceci peut alors avoir des conséquences cliniques importantes chez les patients pouvant aller de la toxicité à la diminution de l'effet thérapeutique.32
1.2.4.2.4 LA SOUS-FAMILLE CYP2E
Cette sous-famille ne contient qu'un seul membre actif soit le CYP2E1. Ce CYP450 ne metabolise spécifiquement que les substrats de petite taille ayant un faible poids moléculaire, dont le chloroforme et le tétrachlorure de carbone. Sa structure ainsi que sa séquence hautement conservées, nous indique que celui-ci joue probablement un rôle physiologique très important. Le gène de ce dernier est localisé sur le chromosome 10q24.3-qter. On retrouve le CYP2E1 principalement au niveau du foie où il occupe 7% du contenu total en CYP450. Il est aussi exprimé dans un grand nombre de tissus incluant les intestins, les leucocytes, le cerveau, la muqueuse nasale et les poumons.18'33"35
1.2.4.3 LA FAMILLE CYP3
Cette famille est considérée comme la plus importante au niveau du foie et de l'intestin. Au niveau du foie celle-ci peut représenter environ de 25 à 28 % des CYP450 totaux et peut même atteindre dans certains cas des taux de 70%. Ce groupe est constitué de 5 gènes CYP3A3, CYP3A4, CYP3A5, CYP3A7 et CYP3A43, tous situés sur le chromosome 7q21-q22.1. Cependant, le CYP3A3 a été abandonné de la nomenclature courante en raison d'évidences grandissantes d'eneurs de séquençage lors d'études antérieures. On remarque que l'expression tissulaire des différents cytochromes diffère : le CYP3A4 est principalement exprimé au niveau hépatique, le CYP3A5 qui est présent
chez environ 15 % de la population est majoritairement exprimé au niveau extra-hépatique, le CYP3A7 au niveau du foie fœtal et le CYP3A43 au niveau de la prostate.
1.2.4.3.1 LECYP3A4
Le CYP3A4 est retrouvé au niveau du foie, où il constitue l'isoforme majeure.36 Il
contribue ainsi à l'effet de premier passage, c'est-à-dire à l'élimination du médicament avant son entrée dans la circulation systémique.37 Le CYP3A4 est aussi le cytochrome
exprimé majoritairement au niveau de l'intestin grêle où il est localisé de façon prédominante au niveau des cellules épithéliales.38' 39 Lown et al. ont déterminé que
l'ADNc du CYP3A4 hépatique et intestinal était identique et que la protéine exprimée dans ces deux tissus était la même. Cependant, il n'existe aucune conélation significative entre l'activité des CYP3A4 hépatiques et intestinaux.40 Le CYP3A4 joue un rôle majeur
dans le métabolisme des médicaments, de ce fait il metabolise plus de 60% des médicaments actuellement sur le marché.41'42 Selon Santé Canada, 22 000 médicaments
seraient actuellement disponibles sur le marché, ce qui souligne l'importance du rôle du CYP3A4 dans le métabolisme de ces derniers. Les médicaments consommés par les patients souffrant de maladies cardiovasculaires sont metabolises par différentes enzymes. Mais plusieurs d'entre eux sont transformés par le CYP3A4, entre autres : les inhibiteurs de l'HMG-CoA reductase, communément appelés statines (atorvastatine, simvastatine, rosuvastatine, etc), les bloquants des canaux calciques (amlodipine, diltiazem, nifedipine, verapamil, etc) et certains anti-arythmiques (quinidine et amiodarone). Plusieurs autres classes de médicaments sont également métabolisées par le CYP3A4 : les anxiolytiques (diazepam, midazolam, alprazolam, etc.), les antidépresseurs
(sertraline, amitryptilline, trazodone, etc), la thérapie contre le VIH (delavirdine, indinavir, zidovudine, etc), les immunosuppresseurs (cyclosporine, tacrolimus), les antipsychotiques (pimozide, clozapine) pour ne nommer que ceux-ci. La grande différence interindividuelle dans l'expression du CYP3A4 n'est pas encore très bien comprise, mais amène une grande variabilité dans le métabolisme et la réponse pharmacologique des médicaments.37' 43' ** Certains prétendent que la variabilité
1.2.4.3.1.1 LA STRUCTURE DU CYP3A4
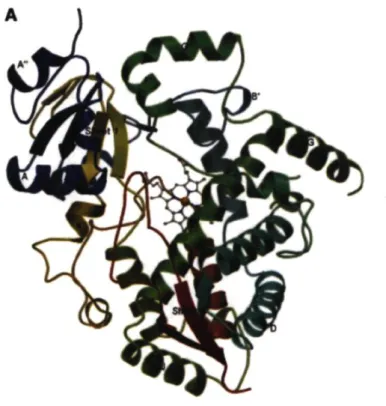
La structure globale tridimensionnelle du CYP3A4 respecte entièrement la conformation caractéristique de l'ensemble des CYP450. Cette structure est représentée ci-bas à la figure 5 et illustre en bleu la partie N-terminale, suivi du vert, suivi du jaune et finissant par le rouge représentant la zone C-terminale de la protéine.
Figure 5. Structure tridimensionnelle du CYP3A4 et de son site actif 46
Tous les CYP450 incluant le CYP3A4 ont une structure protéinique secondaire principalement composé de feuillets-(3 et d'hélice-a. La structure de la région N-terminale composée d'acides aminés hydrophobes forment un domaine riche en feuillets P et constituent la partie transmembranaire de la protéine. Tandis que la structure de la
région C-terminale est caractérisée par un domaine riche en hélice-a formant le site actif et Thème.46
La structure du site actif de 520 Â3 du CYP3A4 permet une liaison avec une
multitude de substrats diversifiés. Cette zone maniable et adaptative peut permette une liaison simultanée de plusieurs substrats, une réalité rendue possible grâce aux différentes conformations du CYP3A4. Plusieurs éléments peuvent jouer sur la conformation de la protéine : la nature du substrat, des facteurs externes tels que le cytochrome b5 et la présence des phospholipides qui peuvent stabiliser certaines conformations. Les zones d'ancrage sont comprises dans les régions entre les hélices B'-C, l'hélice centrale I (en vert au centre), le baneau 1 du feuillet P en N-terminal (en jaune), la poche cysteine ainsi que l'hélice amino-terminale L (en rouge en bas au centre). La poche cysteine comprend 6 résidus conservés dont la cysteine proximale qui se lie au fer tout juste avant le début de l'hélice amino terminale L (en rouge en bas au centre). De ces résidus conservés, c'est le résidu cysteine Cys442 qui joue le rôle de former la liaison thiolate pour le CYP3A4. Toujours pour le CYP3A4, les propionates de l'hème forment des liaisons non-covalentes avec les chaînes latérales des acides aminés Argl05, Trpl26, ArgBO, Arg375 et PArg440.
1.2.4.3.1.2 POLYMORPHISME ET VARIANCE GÉNÉTIQUE
Le cytochrome P450 3A4 est une enzyme fortement conservée à travers les différents individus et les différentes espèces. A ce jour, les différentes recherches n'ont pas pu démontrer l'existence d'un variant fonctionnel de ce gène. Cependant, l'activité interindividuelle de cette enzyme peut être variable, ce qui est encore inexpliqué
aujourd'hui. 4? L'hypothèse est que la variabilité de cette activité soit expliquée par la
modulation de son expression plutôt que par la modification de sa structure. De plus, certains spécialistes affirment que l'existence d'un métabolisateur lent de ce cytochrome est impossible et incompatible avec la vie. Jusqu'à tout récemment, aucun polymorphisme du cytochrome P450 3A4 n'avait été relié à un effet clinique direct. Toutefois une équipe de recherche japonaise a publié en 2007 un article démontrant que le polymorphisme 13989 A-»G (Uell8Val) du cytochrome P450 3A4 était significativement relié au diabète de type IL 48
1.2.4.3.1.3 IMPLICA TION ET RÔLE ENDOGÈNE DU CYP3A4
Comme tous les autres CYP450, le cytochrome P450 3A4 possède un rôle endogène bien défini. Celui-ci participera dans la majorité des cas à des réactions anaboliques du métabolisme. En effet, il participera par exemple à la synthèse des acides biliaires et de la vitamine D. De plus, cette enzyme possède comme substrats endogènes les stéroïdes naturels tels que la testosterone et la progestérone, l'acide arachidonique et le cholestérol.2'49
1.2.4.3.2 LECYP3A5
La séquence d'ADNc du CYP3A5 est à 88% similaire à celle du CYP3A4.50 Les
substrats du CYP3A5 sont en général similaires à ceux du CYP3A4.50 Cette enzyme se
retrouve au niveau du foie, de l'intestin grêle, du sang, des reins, des poumons, de la prostate et de l'hypophyse. '5 2 L'activité polymorphique du CYP3A5, est relativement
variabilité interindividuelle dans le métabolisme des médicaments par la sous-famille CYP3A.53 De plus, une étude in vivo chez l'humain de He et al., démontre que les
variants génétiques identifiés dans le CYP3A4 et le CYP3A5 auraient seulement un faible impact clinique sur le métabolisme par cette sous-famille.54
1.2.4.3.3 LECYP3A7
Le CYP3A7 a quant à lui une séquence d'acides aminés similaire à 88% de celle du CYP3A4.55 Ce dernier est retrouvé surtout chez l'embryon, le fœtus et les
nouveau-nés.56' 57 Son activité atteindrait son maximum dans la première semaine de vie pour
ensuite décliner et atteindre un très faible niveau chez l'adulte.
1.2.4.3.4 LECYP3A43
La séquence d'acides aminés du CYP3A43 serait similaire à 76% à celle du CYP3A4. On le retrouve surtout au niveau de la prostate et des testicules et l'implication de cette protéine au niveau du métabolisme serait faible.59
1.2.4.3.5 LE CYP3A4 ET LA P-GLYCOPROTÉINE (P-GP)
Le métabolisme par les CYP450 et le transport des médicaments par la P-gp doivent être considérés ensemble, afin de prédire avec plus d'exactitude la disposition du médicament chez l'humain.60 Dû à la similitude de leurs substrats, le CYP3A4 et la P-gp
La P-gp et le CYP3A4 au niveau de l'intestin grêle contribuent significativement à une faible biodisponibilité orale et à une variabilité pharmacocinétique des médicaments.
L'expression des cytochromes dans différentes conditions a été étudiée, comme par exemple les maladies du foie, les infections, les maladies de la glande thyroïde, la thérapie avec des hormones de croissance et de la glande hypophysaire et le cancer.63
Certaines de ces conditions augmentent ou diminuent l'expression du CYP3A4.64'65 Des
résultats similaires sont obtenus lors des études portant sur l'expression de la protéine de transport P-gp.66'67 L'interaction fonctionnelle entre ces deux protéines n'est pas très bien
comprise, mais certaines évidences expérimentales suggèrent quelques mécanismes. Leur expression serait co-régulée par les récepteurs nucléaires SXR /PXR.68 La P-gp a pour
rôle de faire sortir le médicament de la cellule, ce dernier peut donc subir une deuxième absorption à l'intérieur de la cellule, il est donc constamment exposé au CYP3A4 ce qui augmente la probabilité que ce dernier soit métabolisé. La P-gp garde une concentration intracellulaire de médicament en relation avec la gamme de concentrations que le cytochrome est capable de métaboliser, c'est-à-dire dans une gamme linéaire de concentrations. Ces deux protéines ont en commun plusieurs substrats, inhibiteurs et inducteurs. Pour éviter une interaction compétitive entre la molécule mère et son metabolite, la P-gp aura une affinité différente pour les deux molécules. Dans le cas où la molécule mère posséderait une plus grande affinité que son metabolite, celui-ci aura alors une plus grande affinité pour un autre système de transport.62'69'70
1.2.5 RÉGULATION DE LA SYNTHÈSE ET DE L 'EXPRESSION DU CYTOCHROME P450 3A4
1.2.5.1 INTRODUCTION SUR L 'INDUCTION ET FACTEURS TRANSCRIPTIONNELS
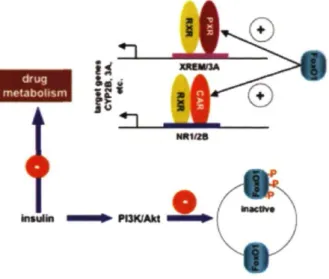
La concentration de plusieurs cytochromes P450 peut être influencée par l'activité de certains récepteurs nucléaires. De plus, ceux-ci sont affectés par une panoplie d'éléments physiologiques et environnementaux.
En effet, un état de stress, l'inflammation, l'âge, le stress oxydatif et plusieurs physiopathologies peuvent interagir avec les récepteurs nucléaires responsables de l'induction des CYP450 (Figure 6). Ceux-ci ont comme propriété d'élever les concentrations des CYP450 en activant les facteurs de transcription cellulaire. La plupart du temps, ces inducteurs sont des substrats de l'enzyme, démontrant un mécanisme d'amplification pour permettre une détoxication plus rapide du corps, en favorisant une augmentation de la vitesse de biotransformation.
La modulation de l'expression des CYP450 par les récepteurs nucléaires peut se traduire par une diminution de l'effet si les metabolites sont inactifs, par une augmentation de l'effet si les metabolites sont actifs ou bien par une augmentation de la toxicité si les metabolites sont réactifs.
Figure 6. Facteurs pouvant influencer l'activité des récepteurs nucléaires Xénobiotiques Hypoxie Grossesse Stress oxydatif Infection Croissance Polluant Inflammation Maladie Polymorphisme Autres Xénobiotique Elimination
L'induction d'une enzyme nécessite plusieurs jours voire semaines avant d'être effective. Comme l'illustre le tableau 3, les substances inductrices du cytochrome P450 les plus connues sont le phénobarbital, la carbamazépine, la rifampicine et l'alcool. Etant donné l'importance des CYP450 dans la biotransformation de nombreux composés, il devient alors important de connaître la régulation de la transcription des CYP450, ainsi que les principales caractéristiques des facteurs de transcription.
Tableau 3 : Enumeration des divers inducteurs du cytochrome P450 3A4 18, 71
Inducteurs du CYP3A4 à l'origine d'interactions médicamenteuses
Antiviraux
éfavirenz, névirapine, Anti-épileptiques
carbamazépine, phénobarbital, phénytoïne, Divers
pioglitazone, rifampicine, bosentan, millepertuis (St. John's wort)
1.2.5.2 LA SUPERFAMILLE DES RÉCEPTEURS NUCLÉAIRES
La famille des récepteurs nucléaires (RN) regroupe plusieurs facteurs de transcription qui sont responsables de l'activation de plusieurs gènes cibles. Pour être activés, ceux-ci doivent se lier à un ligand spécifique et parfois la participation de certains co-facteurs sera nécessaire. Ce complexe alors formé pouna alors se lier à l'ADN et permettre la transcription d'un gène. Le génome humain compte aujourd'hui plus de 48 gènes permettant l'expression des RNs.72 Tous les membres de cette superfamille
possèdent entre eux une structure semblable qui implique la présence de quatre domaines principaux. D'abord une région N-terminale variable, ensuite vient le domaine de liaison à l'ADN (DBD : DNA binding domain) une séquence très conservée, suivi d'une zone variable et de la région C-terminale, siège du domaine de liaison du ligand (LBD : Ligand Binding Domain). Le tableau 4 fait mention des quelques membres d'intérêt de cette famille qui seront discutés plus en détail dans les prochains paragraphes.
Tableau 4 : Récepteurs nucléaires impliqués dans la modulation de l'expression du cytochrome P450 3A4 73-77
Noms Nomenclature Ligands
PXR NR1|2 Xénobiotiques et 16 alpha-cyanopregnenolone
CAR NR1|3 Xénobiotiques et phénobarbital
1.2.5.2.1 LE RÉCEPTEUR NUCLÉAIRE PXR
L'induction du CYP3A4 par les xénobiotiques est largement étudiée depuis plusieurs années. Celle-ci serait due à l'activation d'un récepteur nucléaire orphelin xénosensible découvert chez la souris par trois groupes différents en 1998. C'est le groupe GlaxoSmithKline qui a baptisé ce récepteur «pregnane X receptor» (PXR, NR1I2). PXR est un membre de la superfamille des récepteurs nucléaires. Il possède de très fortes similitudes avec le récepteur de la vitamine D (VDR, vitamin D receptor) et tout comme ce récepteur, celui-ci se lie à l'ADN sous forme d'un hétérodimère en partenariat avec RXR alpha (Retinoid X receptor alpha). Les études de cristallographie ont pu démontrer que le site de liaison du ligand permet selon sa conformation d'accommoder une grande variabilité de structure et de grosseur de ligands différents comparativement aux autres récepteurs nucléaires. Il est alors considéré comme un récepteur non-spécifique ne possédant pas de ligand bien défini. La cible de celui-ci ne se limite pas seulement au cytochrome 3A4, il permet l'induction aussi des familles CYP2B et CYP2C et de la P-glycoprotéine. De plus, PXR se retrouve seulement au niveau du noyau de la cellule. Chez toutes les espèces, PXR est fortement exprimé au foie, cependant il se retrouve à un degré moindre au niveau de l'intestin grêle. Il existe cependant une variabilité inter espèce au niveau de l'induction du CYP3A, cela serait dû aux interactions des composés avec le domaine de liaison du ligand de PXR qui diffèrent d'une espèce à l'autre.49
1.2.5.2.2 LE RÉCEPTEUR NUCLÉAIRE CAR
Dans les débuts de la recherche, on avait observé qu'un traitement au phénobarbital (PB) chez des rats avait comme conséquence directe d'augmenter le métabolisme de certaines drogues. Ce n'est que plusieurs années plus tard qu'il a été possible de démontrer le pouvoir inducteur des PBs sur les cytochromes P450 et plus précisément sur la sous-famille CYP2B. Cette induction est alors expliquée par le pouvoir du phénobarbital à activer le récepteur nucléaire CAR. Tout comme PXR, CAR est un membre de la superfamille des récepteurs nucléaires qui se lie à l'ADN sous forme d'un hétérodimère en partenariat avec RXR alpha (Retinoid X receptor alpha). Ce récepteur fut découvert en 1994 par le groupe de Baes. À cette époque le nom utilisé pour identifier CAR était le MB67.78
1.2.5.2.3 INTERACTION ENTRE PXR ET CAR
La littérature a démontré ces dernières années, qu'il existe bien une forme d'interaction entre CAR et PXR. En effet, il est maintenant établi que CAR peut avoir une influence directe sur l'activation des gènes codant pour la famille CYP3A via son interaction avec PXR. Cette interaction serait à la base de l'explication de l'induction du CYP3A par le phénobarbital.49 De plus, il a été prouvé que PXR possède aussi la
capacité d'activer les gènes codant pour la famille CYP2B par son interaction avec CAR. Cette activation croisée entre ces deux récepteurs nucléaires suggère une protection
supplémentaire dont s'est dotée l'oraganisme pour contrer l'accumulation des composés néfastes au bon fonctionnement du système.4 À titre indicatif, la figure 7 démontre
d'autres interactions existantes faisant varier l'activité des récepteurs nucléaires discutés ci-haut.
Figure 7. Autres interactions possibles pouvant influencer l'activité des récepteurs nucléaires CAR et PXR
C H A P I T R E 2 - I N T R O D U C T I O N : L E S Y N D R O M E M É T A B O L I Q U E
2.1 INTRODUCTION
2.1.1 DÉFINITIONS ET CRITÈRES DIAGNOSTIQUES
La modification des habitudes de vie par une alimentation malsaine ou même par un régime de vie sédentaire a eu comme conséquence une augmentation significative de la progression de l'obésité dans les dernières décennies. Depuis les 25 dernières années, le nombre de Canadiens affichant un surpoids ou une obésité ne cesse de croître. Selon les résultats de l'Enquête sur la santé dans les collectivités canadiennes réalisée en 2004, près du quart (23%) des adultes étaient obèses, c'est-à-dire que leur indice de masse corporelle (IMC) était supérieur à 30 kg/m2, alors qu'en 1978-1979 ce taux d'obésité se
chiffrait à 13,8%. Toujours en 2004, 18% des enfants et adolescents âgés entre 12 et 17 ans affichaient un surpoids et 8% étaient obèses tandis que ces chiffres étaient respectivement 12% et 3% en 1978-1979.79
Ce fléau a ainsi eu comme répercussion une élévation de la survenue de maladies non spécifiques particulièrement d'origine métabolique. Un de ces principaux désordres métaboliques, mieux connu sous le nom de syndrome métabolique, se définit selon le rapport du « National Cholesterol Education Program's Adult Treatment Panel III (ATPIII) » comme un ensemble de marqueurs de risque qui prédisposent aux maladies cardiovasculaires. Dans ce même rapport, on fait mention de 6 critères généraux présents dans le syndrome métabolique et ayant une forte conélation avec l'apparition d'événements cardiovasculaires. Parmi ces critères nous retrouvons : l'obésité viscérale, la dyslipidémie, l'hypertension artérielle, la résistance à l'insuline, les états pro-inflammatoires et l'état pro-thrombotique. Le syndrome métabolique constitue alors un
stade précoce commun à plusieurs maladies majeures, dont le diabète du type II, les maladies cardiovasculaires ainsi que les accidents vasculaires cérébraux (AVC). Afin d'assurer une sensibilisation des professionnels de la santé à ce sujet afin qu'ils puissent porter un diagnostic précoce, certaines publications proposent des critères diagnostiques. En effet, voici un tableau résumé (5) des critères proposés par 1' «ATPIII» et l'Organisation mondiale de la santé (OMS).80
Tableau 5. Listes des divers critères énoncés par V «ATPIII» et l'Organisation mondiale de la santé (OMS) pour émettre un diagnostic de syndrome métabolique '
1. ATPIII (Au moins 3 critères) 2. OMS
Un tour de taille plus élevé que 102 cm chez les • Diabète/résistance à l'insuline hommes et 88 cm chez les femmes.
TG > 1,7 mmol/L Plus 2 des critères suivants :
Faible HDL-C < 1,0 mmol/L chez les hommes • Obésité: IMC > 30 ou ratio taille-hanche Homme > 0,9 et < 1,3 mmol/L chez les femmes Femme > 0,85 Une hypertension > 135/85 mmHg • Dyslipidémie: TG > 1,7 mmol/L ou HDL-C Homme > 0,9 Une glycémie à jeun > 6,1 mmol/L Femme > 0,85
• Microalbinurie > 20fig/min • Hypertension > 140/90
Abréviations : IMC, indice de masse corporelle; HDL-C, High-density lipoprotein-cholesterol; TG, triglycérides
De plus, la thérapie médicamenteuse multiple observée chez les patients souffrant du syndrome métabolique et du diabète, souvent de pairs, est pratique commune. De plus, nous pouvons constater que les problèmes liés aux interactions médicamenteuses sont de plus en plus fréquents en clinique. En effet, certaines études révèlent que des effets indésirables importants arrivent chez environ 8% des patients hospitalisés et 0,32% de ces patients en meurent. Nous pouvons aussi rajouter que la classe des médicaments cardiovasculaires est de loin la plus impliquée dans les cas
d'interactions médicamenteuses. 81'82 À noter que le vocable syndrome métabolique se
référera tout au long du mémoire aux termes diabète de type II et obésité à risque.
Il devient donc primordial de comprendre les répercussions engendrées par ces conditions physiopathologiques sur les activités de biotransformation des drogues. Il est aussi important d'investiguer leurs impacts au niveau des enzymes responsables de la biotransformation des médicaments, afin d'évaluer les possibles interactions médicamenteuses et la survenue d'événements indésirables auprès de la population ayant développé un syndrome métabolique. De plus, étant donné que ces patients ont souvent recours à une thérapie médicamenteuse multiple, il est d'un grand intérêt de vérifier si leur état physiopathologique influence la demi-vie d'élimination de certains médicaments, suggérant alors la nécessité d'un suivi thérapeutique adapté pour cette clientèle.
2.1.1.1 L 'OBÉSITÉ À RISQUE
Le changement des habitudes de vie causé par l'environnement moderne des dernières années permet un accès illimité à des aliments très riches en gras et en sucres sans avoir à dépenser beaucoup d'énergie. Cet environnement qualifié d'«obésogène» influe également sur la pratique de l'activité physique, sa diminution actuelle représentant des risques considérables pour la santé.83
Il est désormais bien documenté que l'obésité à risque porte préjudice à la santé de par sa relation avec le développement de complications métaboliques qui sont à même d'augmenter le risque de développer une maladie cardiovasculaire (MCV) et le diabète de type II. L'obésité augmente aussi l'incidence d'autres facteurs de risque associés aux MCV tels que l'hypertension, la dyslipidémie et un état inflammatoire et pro-thrombotique.
L'expression des CYP450 est beaucoup influencée par notre environnement. Au cours des dernières années, plusieurs études ont mis en évidence l'implication de plusieurs facteurs pouvant affecter l'activité de ceux-ci.
2.1.1.1.1 INFL UENCE DE L 'OBÉSITÉ À RISQUE SUR L 'EXPRESSION DES CYP450
En 1999, les membres d'un groupe de recherche de l'Université de la Caroline du Nord ont effectué une méta-analyse dont l'objectif principal était de vérifier et d'évaluer l'effet de l'obésité sur l'expression des cytochromes P450 3A4, 2E1, 1A2, 2C9, 2C19 et 2D6. Pour ce faire, une revue de la littérature a été effectuée sur l'ensemble des articles parus entre 1966 et 1998 portant sur l'étude comparative de la pharmacocinétique entre des groupes de sujets obèses et non obèses. L'étude a pu démontrer une diminution significative de la biotransformation des substrats du CYP3A4 chez les patients obèses et une augmentation significative de la biotransformation des substrats du CYP2E1 chez ces mêmes sujets. Cependant, les critères d'inclusion de ces analyses se basaient tout simplement sur l'indice de poids corporel des sujets. Cette étude ne fait aucune
discrimination entre les personnes obèses avec syndrome métabolique ou pas. Egalement, à l'instar de cette épidémie d'obésité, une autre maladie ne cesse de progresser, soit le diabète de type II. M
2.1.1.2 LE DIABÈTE
Le diabète est une maladie chronique commune qui depuis quelques années augmente en incidence et ce mondialement. Cette affection est caractérisée comme une maladie multi-systémique menant à des altérations biochimiques et structurelles. Le diabète fait partie d'un des critères diagnostiques du syndrome métabolique. Les conséquences se traduisent par la présence d'une hyperglycémie due à une déficience dans la sécrétion et/ou l'action de l'insuline.85 Il existe deux catégories de diabètes qui se
distinguent entre elles par certaines caractéristiques.
Le diabète de type I est décrit par une réponse immune de la destruction des cellules bêta du pancréas, un processus qui est probablement initié par des facteurs environnementaux chez les personnes prédisposées.86"88
Le diabète de type II est quant à lui caractérisé par une résistance à l'insuline et un défaut dans la sécrétion de cette dernière. En effet, l'installation progressive de la résistance à l'insuline débute souvent avec une prise de poids corporel (figure 8). Il est important de préciser qu'à ce stade, une simple modification des habitudes de vie peut renverser le processus de résistance. Dans cette phase, le corps est capable de contrer
cette résistance par une augmentation de la sécrétion d'insuline. La personne devient alors dans un état d'hyperinsulinémie. Face à cette résistance en pleine progression, le pancréas s'épuisera et sa capacité à sécréter de l'insuline diminuera. À ce moment le corps n'est plus capable de contrebalancer cette résistance et l'hyperglycémie fait son apparition. Tout aussi longtemps que le pancréas est capable de sécréter suffisamment d'insuline, les patients peuvent être traités par une amélioration des habitudes de vie et par l'introduction d'agents pharmacologiques hypoglycémiants. Ces produits auront pour effet d'augmenter la sécrétion d'insuline ou bien de diminuer la néoglucogénèse ou bien de diminuer la résistance à l'insuline. Advenant que le pancréas ne soit plus capable de sécréter de l'insuline, les personnes deviendront alors insulinodépendantes et devront s'auto-injecter de l'insuline recombinante par voie sous-cutanée.
Figure 8. Évolution de l'état pathologique du diabète de type I I 89
. 2 -<û -') ■— u c u e-, •o ■r, C O _o U 4 -o 53 -U D. 2 c o u —_— u 3 ■— r 3 r O 0 . — -o OL. t Pris» y ^ 4e pcijJs /
1
y ^ " N
I n . l t M r f o t t t o M . t Pris» y ^ 4e pcijJs /1
y ^ " N
y I l l U l l i M Capacité i t j.crétion/ t
100 X Apparitlta 4t l'k|r«rfli|céi_ie \ Nécessité d'une \ iM.liMtkérapit ox U l Diététique Can priâtes Ineiulinr Temps (années)Nous pouvons constater que la prévalence du diabète de type II a augmenté de façon alarmante au Canada et aux États-Unis, depuis les dernières années. Ce phénomène
est parfaitement conélé avec l'augmentation de l'obésité et la présence de l'inactivité physique dans nos sociétés modernes. L'association américaine du diabète (ADA) estime que plus de 23 millions d'adultes américains âgés de 20 ans et plus sont atteints de diabète. On estime que plus de 95 % des cas se réfèrent à un diabète de type II. L'Organisation mondiale de la santé (OMS) a estimé qu'environ 170 millions de personnes dans le monde étaient atteintes du diabète et que ce chiffre devrait atteindre approximativement les 366 millions en 2030 (figure 9). En 2005, l'OMS a estimé que 1,6 milliard d'adultes dans le monde souffraient d'embonpoint et que 400 millions souffraient d'obésité, ce qui conèle très bien avec le risque accru de développer des troubles d'hyperglycémie. 90"92
Figure 9. Estimation du nombre d'adultes atteints de diabète par groupe d'âge pour les années 2000 et 2030 dans le monde. D'après Wild et al., 2008. 92
Q.
2L
c
C.2
o =
ci
_o </> £T
§ 1
If
*_• ~ IA LU 200 180160
140120
100
80 60 40 20 0World
ID2000
■ 2030
20-44 45-64Age group (years)
Le diabète de type II est une maladie qui entraîne d'importants troubles associés augmentant ainsi la morbité chez cette clientèle. Ces troubles sont souvent d'origine micro-vasculaire (neuropathie, néphropathie et rétinopathie) et d'origine macrovasculaire (AVC et athérosclérose).
2.1.1.2.1 L'INFL UENCE DU DIABETE SUR L'A CTIVITE ET L'INDUCTION DU P450 CYP3A4
2.1.1.2.1.1 LES ÉTUDES CLINIQUES
Très peu d'études cliniques ont été menées sur l'influence du diabète de type II sur la biotransformation des médicaments. Toutefois, en 2002, une équipe de recherche du Brésil a mené une étude comparative sur la cinétique de la nisoldipine (substrat du CYP3A4) chez deux groupes de sujets hypertendus avec et sans diabète type II. Les données finales de cette étude clinique permettent de conclure à une altération significative de la cinétique de la nisoldipine chez les sujets hypertendus avec diabète. Par conséquent, on peut alors supposer que l'état pathologique du diabète entraînerait une diminution marquée de l'activité du cytochrome P450 3A4.93
2.1.1.2.1.2 LES ETUDES FONDAMENTALES 2.1.1.2.1.2.1 LES ÉTUDES ANIMALES
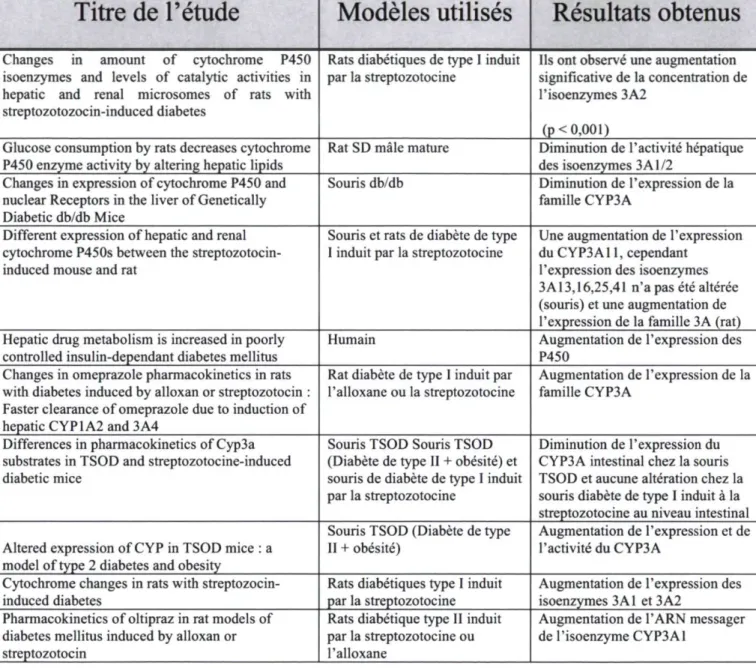
Avec la venue des modèles d'animaux diabétiques de types I et de type II, quelques équipes de recherche ont tenté de caractériser, dans ces états pathologiques, l'expression et l'activité des CYP3A4 hépatiques et intestinaux. Le tableau 6, illustre un résumé de l'ensemble des études identifiées par le moteur de recherche pubmed. Nous constatons rapidement que les études n'arrivent pas à un consensus. Cette divergence dans les résultats laisse planer un doute sur l'influence réelle du diabète sur la biotransformation des médicaments. Dans ces études, deux espèces animales sont utilisées : le rat et la souris. De plus, on remarque que les modèles de diabète de types I sont obtenus grâce à l'utilisation de la streptozotozine et l'alloxane et que les modèles de diabète de type II sont des animaux transgéniques (db/db, ob/ob, TSOD).
Tableau 6. Ensemble des articles obtenus lors d'une revue de littérature portant sur l'influence du diabète sur l'expression du P4S0 CYP3A4rM'm
Titre de l'étude
Modèles utilisés
Résultats obtenus
Changes in amount of cytochrome P450 isoenzymes and levels of catalytic activities in hepatic and renal microsomes of rats with streptozotozocin-induced diabetes
Rats diabétiques de type I induit
par la streptozotocine significative de la concentration de Ils ont observé une augmentation l'isoenzymes 3A2
(p< 0,001) Glucose consumption by rats decreases cytochrome
P450 enzyme activity by altering hepatic lipids Rat SD mâle mature des isoenzymes 3 A1/2 Diminution de l'activité hépatique Changes in expression of cytochrome P450 and
nuclear Receptors in the liver of Genetically Diabetic db/db Mice
Souris db/db Diminution de l'expression de la famille CYP3A
Different expression of hepatic and renal cytochrome P450s between the streptozotocin-induced mouse and rat
Souris et rats de diabète de type
I induit par la streptozotocine Une augmentation de l'expression du CYP3A11, cependant l'expression des isoenzymes 3A13,16,25,41 n'a pas été altérée (souris) et une augmentation de l'expression de la famille 3A (rat) Hepatic drug metabolism is increased in poorly
controlled insulin-dependant diabetes mellitus Humain Augmentation de l'expression des P450 Changes in omeprazole pharmacokinetics in rats
with diabetes induced by alloxan or streptozotocin : Faster clearance of omeprazole due to induction of hepatic CYP1A2 and 3A4
Rat diabète de type I induit par
l'alloxane ou la streptozotocine Augmentation de l'expression de la famille CYP3A Differences in pharmacokinetics of Cyp3a
substrates in TSOD and streptozotocine-induced diabetic mice
Souris TSOD Souris TSOD (Diabète de type II + obésité) et souris de diabète de type I induit par la streptozotocine
Diminution de l'expression du CYP3A intestinal chez la souris TSOD et aucune altération chez la souris diabète de type I induit à la streptozotocine au niveau intestinal Altered expression of CYP in TSOD mice : a
model of type 2 diabetes and obesity
Souris TSOD (Diabète de type II + obésité)
Augmentation de l'expression et de l'activité du CYP3A
Cytochrome changes in rats with
streptozocin-induced diabetes Rats diabétiques type I induit par la streptozotocine Augmentation de l'expression des isoenzymes 3A1 et 3A2 Pharmacokinetics of oltipraz in rat models of
diabetes mellitus induced by alloxan or streptozotocin
Rats diabétique type II induit par la streptozotocine ou l'alloxane
Augmentation de l'ARN messager de l'isoenzyme CYP3A1