Activation du lien C-F de benzyles monofluorés par
utilisation de donneurs de liaisons hydrogène
Mémoire
Camille Houle
Maîtrise en chimie - avec mémoire
Maître ès sciences (M. Sc.)
Activation du lien C
‒F de benzyles monofluorés par utilisation de
donneurs de liaisons hydrogène
Mémoire
Camille Houle
Sous la direction de :
Résumé
Depuis quelques décennies, on constate un intérêt marqué pour la synthèse de composés organofluorés en chimie organique. En effet, la grande stabilité de ceux-ci, principalement expliquée par la grande force du lien C‒F, en font des composés attirant dans diverses sphères de la chimie telles la chimie pharmaceutique, l’agrochimie et la chimie des matériaux.
Étant donné la force de la liaison C‒F, de l’ordre de 450 kJ/mol, et sachant que le fluorure est un mauvais groupe partant, il peut sembler peu intuitif de faire réagir cette liaison. Pourtant, certains chercheurs s’intéressent particulièrement au clivage du lien C‒ F par diverses méthodes afin de fonctionnaliser cette liaison: on parle alors d’activation de liaisons C‒F.
Parmi les propriétés intéressantes du fluor organique, on compte le fait qu’il est faiblement accepteur de liaisons hydrogène en solution. Dans cet ordre d’idée, le groupe de recherche a développé au fil des années plusieurs méthodologies permettant le départ du fluor dans des composés organiques en utilisant des espèces donneuses de ponts hydrogène.
Le premier projet consiste en l’utilisation d’une thiourée comme composé donneur de liaisons hydrogène afin d’activer la liaison C‒F benzylique. En utilisant une quantité catalytique de thiourée, il a été possible d’effectuer une réaction de type SN2 de différentes amines sur plusieurs fluorures benzyliques. Il s’agit également du premier exemple d’activation organocatalysée par interactions hydrogène de liens C‒F benzyliques.
Le deuxième projet consiste en la valorisation d’une méthode précédemment publiée par le groupe de recherche. En utilisant un mélange de solvant donneurs de liaisons hydrogène pour la réaction, il est possible d’effectuer une réaction SN2 d’une gamme de nucléophiles sur des différents substrats fluorés, et ce, avec de très bons rendements isolés.
Abstract
There has been, in the past decades, a keen interest from the organic chemistry community for the synthesis of organofluorine compounds. Indeed, the high stability of those compounds, mostly explains by the great stability of the C‒F bond, makes them attractive for various chemistry applications, including pharmaceutical, agrochemical and material chemistry.
Because of the C‒F bond strength, which is around 450 kJ/mol, and the poor leaving group ability of fluoride, the reactivity of organofluorine compounds is somewhat limited. That being said, the cleavage of the C‒F bond by various methods, also called C‒ F bond activation, has attracted it share of attention.
One of the various properties of organic fluorides is its ability to accept hydrogen bonds in solution. Along these lines, our research group has developed, over the years, a number of methods to trigger the activation of the C‒F bond using hydrogen bond donor species.
The first project involves the use of a thiourea as a hydrogen bond donor for the activation of benzylic C‒F bonds. Using a catalytic amount of thiourea, it is possible to perform SN2 reactions on various benzylic fluoride using different amines. This method is the first example of benzylic C‒F bonds activation organocatalyzed by hydrogen bond interactions.
The second project focuses on the valorization of previously published method by our research group. Here, using a mixture of hydrogen bond donor solvents, it is possible to conduct SN2 reactions on various fluorinated substrates with a range of nucleophiles with excellent isolated yields.
Table des matières
1
Résumé ... ii
Abstract ... iii
Table des matières... iv
Liste des schémas ... vii
Liste des figures ... ix
Liste des tableaux ... x
Liste des abréviations, sigles, acronymes ... xi
Remerciements ... xiv
Introduction ... 1
Histoire du fluor ... 1
Propriétés de l’atome de fluor ... 2
Composés organofluorés ... 4
Activation de la liaison C‒F ... 6
Méthodes d’activation de fluor aliphatique ... 7
Liaisons hydrogène ... 12
Méthodes d’activation de fluor aliphatique par liaisons hydrogène ... 15
Objectifs ... 19
1 Activation de la liaison C‒F par une thiourée ... 21
1.1 Introduction thiourée ... 21
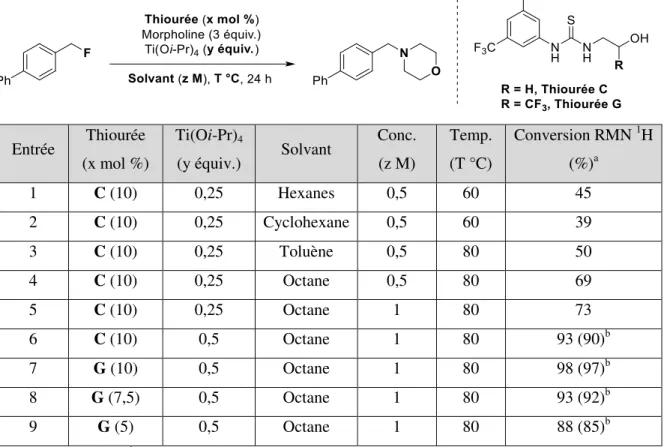
1.2 Résultats préliminaires ... 23
1.2.1 Sélection de la thiourée ... 23
1.2.2 Résultats initiaux version catalytique ... 26
1.3 Optimisation ... 29
1.4 Résultats et discussion ... 32
1.4.1 Nucléophiles aminés ... 33
1.4.2 Nucléophiles oxygénés, soufrés et carbonés ... 38
1.4.3 Substrat de départ ... 40
1.5 Études mécanistiques ... 43
1.6 Conclusion ... 47
1.7 Partie expérimentale ... 47
1.7.1 General information ... 47
1.7.2 Synthesis of the benzyl fluorides ... 48
1.7.3 Synthesis of thioureas catalysts ... 55
1.7.4 Nucleophilic substitution reaction ... 58
2 Activation de la liaison C‒F par un solvant donneur de ponts hydrogène ... 70
2.1 Résultats préliminaires ... 70
2.2 Optimisation ... 72
2.3 Résultats et discussion ... 75
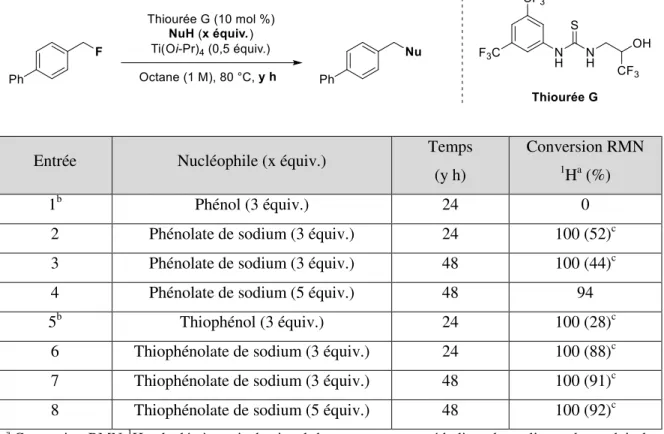
2.3.1 Nucléophiles aminés ... 75
2.3.2 Nucléophiles oxygénés ... 78
2.3.3 Nucléophiles soufrés et carbonés ... 80
2.3.4 Substrats de départ ... 82
2.4 Conclusion ... 83
2.5 Partie expérimentale ... 84
2.5.1 General information ... 84
2.5.2 Synthesis of the benzyl fluorides ... 85
Conclusion ... 99 Travaux futurs ... 100 Bibliographie... 101
Liste des schémas
Schéma i.1 : Réaction de E1cb et de substitution nucléophile aromatique ... 4
Schéma i.2 : Réaction de glycosilation sur le fluorure de glycosyle ... 8
Schéma i.3 : Hydrodéfluoration d'alkyl fluoré par B(C6F5)3 ... 8
Schéma i.4 : Synthèse d'oxazolidinones par activation de lien C‒F ... 9
Schéma i.5 : Formation de lien C‒C à l’aide de Al(C6F5)3 ... 9
Schéma i.6 : Activation de la liaison C‒F par un acide de Brønsted ... 10
Schéma i.7 : Activation de lien C‒F allylique par un catalyseur de palladium ... 11
Schéma i.8 : Activation de lien C‒F benzylique par un catalyseur de palladium ... 11
Schéma i.9 : Activation par une mélange de solvants donneurs de liaisons hydrogène ... 16
Schéma i.10 : Activation par un triol ... 17
Schéma i.11 : SN1 sur des substrats benzyliques par activation avec HFIP ... 18
Schéma i.12 : réaction de SN1 en utilisant une quantité catalytique de TFA pour l'activation ... 18
Schéma i.13 : Projets décrits dans le cadre de ce mémoire ... 20
Schéma 1.1 : Réaction de Diels-Alder catalysée par une thiourée ... 22
Schéma 1.2 : Réaction de Baylis-Hillman asymétrique en utilisant une bis-thiourée chirale ... 23
Schéma 1.3 : Méthode de synthèse des thiourées ... 24
Schéma 1.4 : Résultats d'activation du lien C‒F par différentes thiourées ... 24
Schéma 1.5 : Résultats préliminaires en utilisant une quantité catalytique de thiourée ... 26
Schéma 1.6 : Étendue de la réaction en utilisant différentes amines secondaires comme nucléophile ... 34
Schéma 1.7 : Étendue de la réaction en utilisant des anilines et amine aromatique comme nucléophile ... 36
Schéma 1.8 : Étendue de la réaction en utilisant d'autres amines primaires comme nucléophile ... 37
Schéma 1.9 : Étendue de la réaction en utilisant des nucléophiles oxygénés, soufrés et carbonés ... 40
Schéma 1.10 : Étendue de la réaction en utilisant d'autres substrats de départ benzyliques ... 42
Schéma 1.11 : Étendue de la réaction en utilisant d'autres substrats de départ fluorés .... 43 Schéma 1.12 : Résultats de l'étude mécanistique de la réaction ... 46 Schéma 2.1 : Étendue de la réaction précédemment publiée par le groupe de recherche 71 Schéma 2.2 : Étendue de la réaction en utilisant diverses amines secondaires comme nucléophile ... 76 Schéma 2.3 : Étendue de la réaction en utilisant diverses amines primaires comme nucléophile ... 78 Schéma 2.4 : Étendue de la réaction en utilisant divers phénols comme nucléophile ... 80 Schéma 2.5 : Étendue de la réaction en utilisant des nucléophiles soufrés et carbonés ... 82 Schéma 2.6 : Étendu de la réaction en changeant le fluorure benzylique de départ ... 83 Schéma c.1 : Résumé du premier projet ... 99 Schéma c.2 : Résumé du deuxième projet ... 100
Liste des figures
Figure i.1 : Composés fluorés utilisés en industrie chimique ... 6
Figure i.2 : Hybridation du fluor dans les composés monofluorés ... 7
Figure 1.1 : Exemples de fonctions chimiques utilisées comme organocatalyseurs ... 21
Figure 1.2 : Thiourée empoisonnée par le fluorure éjecté ... 27
Figure 1.3 : Produits observés par RMN 1H lors du test contrôle à 100 °C sans réactifs . 31 Figure 1.4 : Possible produit secondaire observé lors de la synthèse du composé 20 ... 37
Liste des tableaux
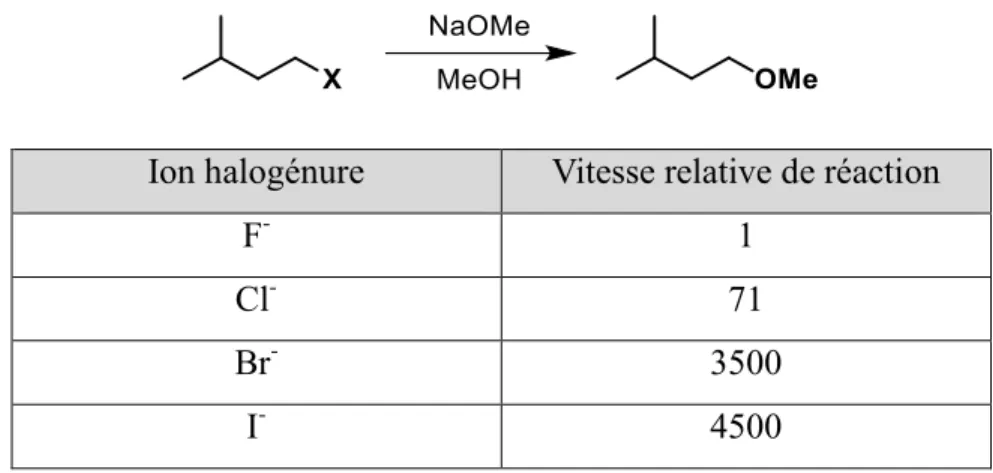
Tableau i.1 : Vitesse de SN2 sur différents halogènes... 3
Tableau i.2 : Force de la liaison CH3‒X en kJ/mol... 4
Tableau i.3 : Énergie calculée de quelques liaisons hydrogène ... 15
Tableau 1.1 : Résultats obtenus lors de la sélection du capteur ... 28
Tableau 1.2 : Optimisation de la réaction d’activation par organocatalyse avec une thiourée ... 30
Tableau 1.3 : Tests contrôles pour l'activation organocatalysée par une thiourée ... 31
Tableau 1.4 : Optimisation des conditions en utilisant le phénol ou le thiophénol comme nucléophile ... 39
Liste des abréviations, sigles, acronymes
Ac Acétyle
Ar Aryle
Bn Benzyle
Boc tert-Butoxycarbonyle BSA Bis(triméthylsilyl)acétamide Bu Butyle cat. Catalytique CFC Chlorofluorocarbone Conc. Concentration DBU 1,8-Diazabicyclo[5.4.0]undéc-7-ène DCE Dichloroéthane
DFT « Density Functional Theory », Théorie de la Fonctionnelle de la Densité
DMF Diméthylformamide
TFA Acide trifluoroacétique
Et Éthyle
E1cb Élimination unimoléculaire base conjuguée
FLP « Frustrated Lewis Pairs », Paires de Lewis Frustrées HFIP Hexafluoroisopropanol
IR Infrarouge
LA « Lewis Acid », Acide de Lewis
Me Méthyle
MTBE Méthyl-tert-butyléther
Nu Nucléophile
RMN Résonance magnétique nucléaire SN2 Substitution nucléophile bimoléculaire SN1 Substitution nucléophile unimoléculaire TBACN Cyanure de tétrabutylammonium Temp. Température
THF Tétrahydrofurane TMS Triméthylsilyle T. P. Température pièce
À mes parents, Chantal Gingras et Christian Houle, Pour avoir toujours cru en moi
Remerciements
Je tiens tout d’abord à remercier mon directeur de recherche, Jean-François Paquin, pour m’avoir donné l’immense privilège d’effectuer mes études graduées au sein de son laboratoire ainsi que pour ta confiance lors de celles-ci. Ta grande disponibilité, tes réponses rapides aux courriels et ton sens de l’organisation ont été fortement appréciés pendant ces deux dernières années.
J’aimerais également remercier le groupe Paquin 2018-20 dans son entièreté (Jean-Denys, Myriam, Mélissa, Majdouline, Marius, Marie, Raphaël, Xavier, Chloé, Audrey) pour l’aide qu’ils ont apportés à mon projet et pour le plaisir qu’on s’est fait. Chacun à votre manière avez contribué à me faire avancer comme personne et fait en sorte que ces dernières années ont passé si vite. Je tiens particulièrement à remercier Marie, ma complice de « juste une maitrise », pour les réponses à toutes mes questions maitrises, chimiques, etc. mais également pour m’avoir écouté et conseillé lorsque j’en ai eu besoin. Tu es une personne extraordinaire et je suis certaine que tu seras une super prof! Également merci à Xavier et Raph, pour toutes les conversation random et pour m’avoir fait bien rigoler pendant les deux dernières années! Merci à Chloé pour ton immense gentillesse. J’ai adoré discuter avec toi et je compte bien allez te rendre visite en France un de ces jours! Je tiens aussi à remercier Jean-Denys, pour m’avoir appris à être plus à l’aise au laboratoire, et à toutes les personnes qui ont travaillés sur les projets des thiourées. Je ne vous connais pas personnellement, mais votre contribution a permis d’enrichir ce projet et à assurer sa publication.
Parfois, la vie nous amène à quitter les sentiers battus et à s’éloigner de ceux qu’on aime. Je tiens à dire un gros merci à mes parents, sans qui mes études à Québec auraient été impossible sur plusieurs aspects. Merci maman, pour ton écoute active, tes conseils judicieux et pour être autant présente malgré la distance qui nous sépare. Tu es une personne très inspirante pour moi, sache que tu es mon modèle! Merci papa, toi aussi tu m’inspires par ta persévérance et ta débrouillardise. À chaque fois que tu me dis que tu es fière de moi, ça me donne la motivation de continuer. Je suis heureuse d’être votre fille et
je vous aime fort! Merci à ma petite sœur, Audrey, pour tous nos fous rires qui me manquent cruellement. Je pense énormément à toi et je suis tellement fière de toi!
Lors de mon aventure à Québec, j’ai également rencontré une personne qui est devenu très importante pour moi. Je tiens à remercier mon amoureux, Emmanuel Brousseau, qui sait tant me faire rire même dans les moments plus sombres. Merci d’avoir enduré mes humeurs de maitrise et m’avoir soutenu pendant ces dernières années. Je t’ai déjà dit que je t’aime?
Merci encore à vous tous pour avoir été présent dans ma vie lors de mes études graduées. Je réalise aujourd’hui qu’une nouvelle page de ma vie se tourne, et chacun d’entre vous y avez inscrit un paragraphe.
Introduction
Histoire du fluor
En 1809, le chimiste français André-Marie Ampère fut le premier à mentionner l’existence du fluor comme élément. Il émit cette hypothèse en comparant l’acide chlorhydrique (un acide de chlore) et l’acide fluorhydrique, utilisé depuis le 18e siècle pour la gravure du verre. Depuis ce temps, plusieurs se succédèrent pour isoler cet élément, généralement par électrolyse des fluorures de phosphore et d’arsenic.1
Environ 80 ans plus tard, ce fut un autre chimiste français, Henry Moissan, qui isola pour la première fois le fluor sous forme d’un gaz jaune-vert, résolvant ainsi un des grands défis de la chimie inorganique. Pour ce faire, il effectua l’électrolyse d’un mélange de HF et de KF en utilisant des anodes et cathodes en platine et à basse température pour limiter la corrosion. Il obtiendra le prix Nobel de chimie en 1906, notamment pour la création d’un four à arc électrique et pour l’isolation du fluor.1
Quelques décennies après la découverte de Henry Moissan, les composés fluorés commencèrent à faire leur apparition en industrie. Tout débute avec l’utilisation des CFC comme réfrigérants. L’attrait de ces composés de fluors et de chlores vient de leur grande volatilité et faible réactivité. De plus, l’essor de la chimie du fluor a été fortement encouragé par le projet Manhattan avec les développements de la chimie nucléaire. En effet, l’une des méthodes de purification de l’uranium consistait à transformer celui-ci en un gaz, l’hexafluorure d’uranium, en utilisant de l’acide fluorhydrique et du fluor gazeux. Finalement, le fluor émergera également en chimie des matériaux, avec l’invention du Téflon, ainsi qu’en chimie pharmaceutique et en agrochimie.2,3
1 Tressaud, A. Angew. Chem. Int. Ed. 2006, 45, 6792.
2 Kirsch, P. Dans Modern Fluoroorganic Chemistry, 2e éd.; Wiley-VCH: Weinheim, Germany, 2013. 3 Hiyama, T. Dans organofluorine compounds; chemistry and applications; Yamamoto, H., Ed.; Springer:
Propriétés de l
’atome de fluor
Possédant un numéro atomique de 9 et une masse atomique de 19, le fluor (F) est un atome fort intéressant. Faisant partie de la famille des halogènes, le fluor possède cependant une réactivité légèrement différente de ceux-ci étant donné ses propriétés.
D’abord, le fluor est l’atome le plus électronégatif du tableau périodique avec une électronégativité de 3,98 sur l’échelle de Pauling. Il s’agit aussi du deuxième plus petit atome du tableau périodique après l’hydrogène, avec un rayon atomique de 1,47 Å. Ces propriétés font en sorte que le fluor est un atome très peu polarisable, car ses orbitales électroniques sont rapprochées de son noyau. De ce fait, les composés fluorés sont plutôt volatils, puisqu’ils effectuent très peu d’interactions de van der Waals entre eux.2
Aussi, le fluor est souvent comparé à l’atome d’hydrogène par certaines de ses propriétés. D’abord, sa taille est très semblable à celle de l’hydrogène (rayon atomique de 1,20 Å). Ensuite, le lien que le fluor forme avec le carbone est d’une longueur d’environ 1,40 Å, ce qui se compare avec la liaison C‒H qui est d’environ 1,09 Å. Ces propriétés font en sorte qu’il est possible de changer un atome d’hydrogène par un atome de fluor dans une molécule organique sans engendrer de grandes perturbations stériques.2
Finalement, contrairement aux autres halogènes, le fluor est considéré comme un mauvais groupe partant (Tableau i.1). Cela est principalement dû à deux facteurs : la faible polarisabilité de l’atome de fluor et la faible stabilisation de l’ion fluorure en solution. En effet, les stabilisations électrostatiques dues à la grande force du lien C‒F font en sorte qu’une polarisation vers l’éjection d’un fluorure ne sera généralement pas observée.4,5
4 Chambers, R. D., dans Fluorine in Organic Chemistry, Blackwell Publishing Ltd., Oxford, 2004. 5 O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308.
Tableau i.1 : Vitesse de SN2 sur différents halogènes
Ion halogénure Vitesse relative de réaction
F- 1
Cl- 71
Br- 3500
I- 4500
Également, pour qu’un atome soit un bon groupe partant, il faut que l’ion formé lors de la substitution soit stable. Plus le pKa de l’acide conjugué au groupement partant est faible, plus celui-ci est stabilisé. Ainsi, comme l’acide fluorhydrique est un acide faible (pKa de 3,20 dans l’eau) comparé aux autres acides halogénés, l’ion fluorure est moins stabilisé en solution que les autres ions halogénures.4,6
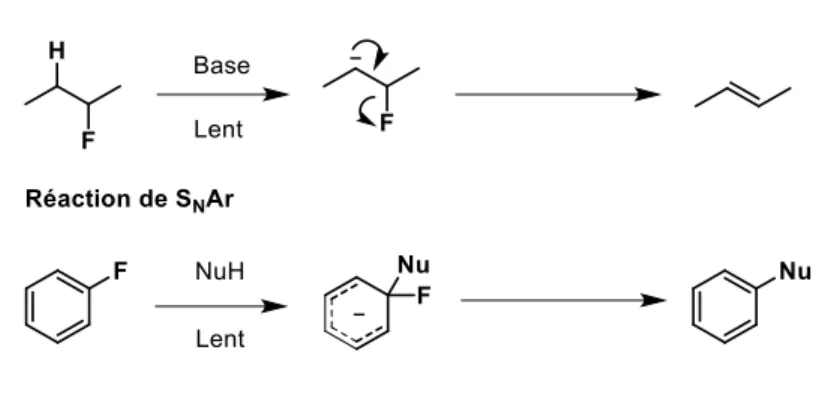
Il existe cependant certaines exceptions où le fluorure peut agir comme groupement partant (Schéma i.1). Dans les réactions d’élimination de type E1cb, le caractère électronégatif du fluor permet de migrer efficacement l’anion pour former une double liaison et éjecter celui-ci. Le même phénomène est observé dans le cas des réactions de substitution nucléophile aromatique ou vinylique, où l’électronégativité du fluor augmente le caractère électrophile du carbone adjacent. Dans ces cas, le fluor est même considéré comme le meilleur groupe partant parmi les halogènes.4
6 (a) Issari, B.; Stirling, C. J. M. J. Chem. Soc., Chem. Commun. 1982, 684. (b) Boyd, D. B. J. Org. Chem.
Schéma i.1 : Réaction de E1cb et de substitution nucléophile aromatique
Composés organofluorés
Lorsqu’introduit dans une molécule organique, le fluor peut venir changer les propriétés de celle-ci. Les caractéristiques que présente la liaison carbone-fluor expliquent les nombreuses applications des composés organofluorés et seront énumérées ci-dessous.
Le lien C‒F est la liaison simple la plus forte que le carbone effectue avec un autre élément. Avec une énergie de liaison de l’ordre de 473 kJ/mol, la liaison C‒F est plus stable que la liaison C‒H ainsi que les liaisons C‒X que forme le carbone avec les autres halogènes (Tableau i.2).2,7 De plus, la stabilité du lien C‒F augmente avec le nombre d’atomes de fluor présents sur le centre carboné tandis que la longueur de la liaison va diminuer. Cela est expliqué par une augmentation du caractère δ+ au niveau du carbone pauvre en électron et donc, une augmentation de l’attraction électrostatique avec les atomes de fluor riches en électrons.5
Tableau i.2 : Force de la liaison CH3‒X en kJ/mol
X H F Cl Br I
Force du lien C‒X (kJ/mol)
440 473 351 292 238
Aussi, étant donné la différence élevée d’électronégativité entre l’atome de fluor et de
7 Uneyama, K. Dans Organofluorine Chemistry, Blackwell Publishing Ltd.; Oxford, Grande-Bretagne,
carbone, la liaison C‒F est très polarisée et porte un certain caractère ionique. Cela vient faire en sorte que le lien C‒F peut effectuer des interactions électrostatiques au sein d’une molécule ce qui en modifie la conformation. Cette grande électronégativité fait également du fluor un groupement électroattracteur, ce qui permet de modifier le pKa des molécules fluorées. La présence d’un atome de fluor sur une molécule vient donc influencer l’acidité ou la basicité de celle-ci ce qui peut modifier, en chimie médicinale par exemple, sa biodisponibilité. Aussi, l’incorporation d’un atome de fluor à un composé vient généralement augmenter la lipophilie de celui-ci lorsque le fluor est aromatique ou adjacent à un système π. Dans ce cas, étant donné le recouvrement des orbitales entre celles du fluor et celles du système π, la liaison C‒F devient très peu polarisée. Au contraire, dans le cas des alkyles monofluorés ou trifluorés, l’atome de fluor vient diminuer la lipophilie, puisqu’il s’agit de groupements polaires.7,8
Ces propriétés du lien C‒F font en sorte qu’un intérêt toujours grandissant est accordé aux composés organofluorés. D’abord, en chimie pharmaceutique, l’introduction d’un fluor à un médicament permet de changer la lipophilie, le pKa, la conformation et la solubilité d’une molécule ainsi que sa stabilité métabolique, étant donné que le fluor est difficilement métabolisé par les cytochromes P450 présents chez l’humain. C’est pourquoi environ 25 % des médicaments en production comportent un ou plusieurs atomes de fluor.8 Un exemple communément mentionné est la fluoxétine (Prozac®) qui comporte un groupement ‒CF3 en para d’un de ses cycles aromatiques. Ces changements qu’apporte l’atome de fluor aux molécules organiques expliquent également la forte présence de celui-ci en agrochimie comme herbicides, fongicides pesticides. D’ailleurs, environ le quart des herbicides licenciés comportent au moins un atome de fluor dont un exemple important est la Florasulam.9 Finalement, le profil électronique du fluor amené par son caractère électronégatif fait en sorte qu’il possède plusieurs applications en chimie des matériaux avec les composés perfluorés (Téflon) et les polymères fluorés (matériaux énergétiques)10 (Figure i.1).
8 Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320.
9 (a) Jeschke, P. ChemBioChem 2004, 5, 570. (b) Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167,
16.
Figure i.1 : Composés fluorés utilisés en industrie chimique
Activation de la liaison C
‒F
L’activation de liens C‒F implique de briser la liaison entre le carbone et le fluor afin d’effectuer de la réactivité au niveau du carbone. D’abord, il s’agit d’un défi intéressant au point de vue de la réactivité. En effet, comme expliqué précédemment, le fluor forme avec le carbone le lien simple le plus fort en chimie organique et l’ion fluorure est considéré comme un mauvais groupe partant étant donné la faible polarisabilité de ce lien et la faible stabilisation du fluorure en solution.2,4,5,6,7 Ensuite, l’activation de liaisons C‒ F permettrait certaines applications potentielles en chimie organique et environnementale.11
L’activation sélective de la liaison C‒F dans des composés ‒CF2 et ‒CF3 comporte également un défi supplémentaire puisque, comme mentionné plus haut, le lien C‒F devient plus fort et plus court lorsqu’on augmente le nombre de fluors présents sur le carbone.5 Cependant, comme les travaux présentés dans ce mémoire se concentrent sur l’activation de benzyles monofluorés, l’accent sera mis sur les méthodes d’activation de composés comportant un seul atome de fluor.
Les composés monofluorés peuvent être séparés en trois catégories : le fluor hybridé sp,
sp2 et sp3 (Figure i.2). Les composés fluorés sp comportent les fluoroalcynes qui sont des composés instables. Pour les composés fluorés sp2, on compte les fluors aromatiques et vinyliques, dont l’activation est bien décrite dans la littérature. Finalement, les composés
organofluorés sp3 comportent les fluors aliphatiques, qui sont plus difficiles à activer que leurs analogues sp2. Les fluorures benzyliques principalement utilisés dans le cadre de ce mémoire font partie de cette catégorie.
Figure i.2 : Hybridation du fluor dans les composés monofluorés
Méthodes d’activation de fluor aliphatique
Dans la littérature, il existe plusieurs méthodes d’activation de liens C(sp3)‒F. Les méthodes les plus communes sont en utilisant des acides de Lewis ou des métaux de transition. Il est également possible d’activer la liaison C‒F en condition basique pour éjecter du HF, mais la majorité des cas décrits montrent que l’élimination se produit via un mécanisme E1cb ou le fluor peut agir comme groupe partant.11,12,13,14 Afin de montrer l’importance de chacune des méthodes, voici quelques exemples.
D’abord, l’utilisation d’acide de Lewis est efficace pour l’activation de liaisons C(sp3)‒F étant donné que celle-ci peut agir comme base de Lewis. L’un des premiers exemples remonte au début des années 80, où Mukaiyama et ses collaborateurs ont montré une méthode très intéressante d’activation en chimie des sucres. Ils ont démontré qu’en utilisant du SnCl4 comme acide de Lewis, la réaction de glycosylation sur le β-fluorure de glycosyle favorisait une sélectivité de l’anomère α pour le produit final.15 (Schéma i.2)
12 Shen, Q.; Huang, Y.-G.; Liu, C.; Xiao, J.-C.; Chen, Q.-Y.; Guo, Y. J. J. Fluorine Chem. 2015, 179, 14. 13 Hamel, J.-D.; Paquin, J.-F. Chem. Commun. 2018, 54, 10224.
14 Stahl, T.; Klare, H. F. T.; Oestrich, M. ACS Catal. 2013, 3, 1578. 15 Mukaiyama, T.; Murai, Y.; Shoda, S. Chem. Lett. 1981, 3, 431.
Schéma i.2 : Réaction de glycosylation sur le fluorure de glycosyle
Plus récemment, le groupe de Stephan a montré une méthode efficace d’hydrodéfluoration d’alkyles fluorés en utilisant un acide de Lewis boré, soit le B(C6H5)3. Il a montré que la réaction se produisait en utilisant une quantité stœchiométrique d’acide et un sel de phosphore et de bore via une réactivité de type FLP. Il a également constaté que la réaction n’avait pas lieu lorsque seul le sel phosphoré et boré était présent dans le milieu, montrant l’importance de l’activation du fluor par le B(C6F5)3. En se basant sur ce constat, le groupe a développé une méthode catalytique de la transformation, en utilisant du triéthylsilane comme source d’hydrure (Schéma i.3). Celle-ci permet l’obtention de rendements presque quantitatifs avec un temps réactionnel de seulement cinq minutes.16
Schéma i.3 : Hydrodéfluoration d'alkyl fluoré par B(C6F5)3
L’activation du lien C(sp3)‒F est bien décrite en utilisant des composés d’aluminium et de silicium étant donné le caractère fluorophile de ceux-ci. Par exemple, Shibata et Haufe ont démontré la synthèse de différents oxazolidinones en utilisant du BSA, un composé silylé, et du fluorure de césium en quantité catalytique (Schéma i.4). Ici, le mécanisme d’éjection du fluor passe par l’interaction avec le groupement ‒OSiMe3 du BSA ce qui
permet l’attaque nucléophile de l’amide préalablement déprotoné. Il s’agit d’une méthode efficace d’activation où le lien C‒F est en position très peu activée.17
Schéma i.4 : Synthèse d'oxazolidinones par activation de lien C‒F
Pour l’activation de liaison C(sp3)‒F en utilisant un composé d’aluminium comme acide Lewis, le groupe de Young a montré la formation d’un lien C‒C en utilisant différents fluorures aliphatiques comme substrats de départ, dont des fluorures benzyliques (Schéma i.5). Dans ce cas, l’activation est sélective à la liaison C‒F (peu de conversion est observée pour l’analogue bromé) et fonctionne même pour les alkyles fluorés peu activés, malgré la difficulté que présente la formation de liens C‒C.18
Schéma i.5 : Formation de lien C‒C à l’aide de Al(C6F5)3
Il est aussi possible d’activer le fluor en condition acide par l’utilisation d’acides de Brønsted forts. Ici, l’activation aurait lieu soit par une forte interaction C‒F…H+ ou par des interactions de type liaisons hydrogène14,19 (détaillé dans la section « liaisons hydrogène » du mémoire). Un récent exemple d’activation par acide de Brønsted a été présenté par Muniz et ses collaborateurs.20 Pour valoriser les produits finaux obtenus, ils ont effectué diverses transformations chimiques sur ceux-ci, dont une défluoration en
17 Haufe, G.; Suzuki, S.; Yasui, H.; Terada, C.; Kitayama, T.; Shiro, M.; Shibata, N. Angew. Chem. Int. Ed.
2012, 51, 12275.
18 Jaiswal, A. K.; Goh, K. K. K.; Sung, S.; Young, R. D. Org. Lett. 2017, 19, 1934.
19 Pour des revues sur la liaison C‒F comme accepteur de liaisons hydrogène, voir : (a) Schneider, H.-J.
Chem. Sci. 2012, 3, 1381. (b) Champagne, P. A.; Desroches, J.; Paquin, J.-F. Synthesis 2015, 306.
utilisant l’acide trifluoroacétique. La réaction de substitution nucléophile a permis d’obtenir l’ester benzylique correspondant via une réactivité intramoléculaire et ce, avec d’excellents rendements (Schéma i.6).
Schéma i.6 : Activation de la liaison C‒F par un acide de Brønsted
Une autre méthode d’activation de lien C(sp3)‒F bien décrite consiste à l’utilisation de métaux de transition. La plupart des cas d’activation à partir de métaux se font sur des substrats benzyliques ou allyliques, puisqu’ils ont la possibilité de former un complexe π-allyle favorable comme montré initialement par le groupe de Hintermann en 2006.21
Gouverneur et Brown ont développé en collaboration plusieurs méthodes d’activation de fluorures benzyliques et allyliques en utilisant des métaux de transition de la 10e famille. Tout d’abord, ils ont montré la réaction de Tsuji-Trost sur des fluorures allyliques en vue de former une liaison C‒C avec un malonate (Schéma i.7). Il est intéressant de noter que dans cette réaction, le fluor est un meilleur groupe partant que l’acétate. Cependant, elle présente certains problèmes de stéréochimie lorsque la réaction est effectuée sur un fluorure allylique énantiopur.22 Alors qu’une double inversion menant à une rétention de configuration serait attendue en utilisant le diméthyle malonate comme nucléophile, le résultat observé est une inversion de configuration montrant des problèmes de sélectivité (syn :anti = 88 : 12). Plus tard, les mêmes auteurs ont montré qu’en changeant le catalyseur de palladium pour un catalyseur de platine, la réaction est alors stéréosélective
21 Hintermann, L.; Läng, F.; Maire, P.; Togni, A. Eur. J. Inorg. Chem. 2006, 1397. 22 Hazari, A.; Gouverneur, V., Brown, J. M. Angew. Chem. Int. Ed. 2009, 48, 1296.
et le fluor est le meilleur groupe partant comparé aux autres fonctions testées (carbonate, acétate, benzoate).23
Schéma i.7 : Activation de lien C‒F allylique par un catalyseur de palladium
Gouverneur et Brown ont également présenté une méthode d’activation de fluorures benzyliques en utilisant un catalyseur de palladium. La méthode est efficace pour la substitution de nucléophiles carbonés, soufrés, oxygénés et aminés sur différents benzyles monofluorés (Schéma i.8). Comme dans le cas d’activation de fluorures allyliques par le palladium, des problèmes de stéréosélectivité sont observés lorsqu’un fluorure benzylique énantioenrichi est utilisé.24
Schéma i.8 : Activation de lien C‒F benzylique par un catalyseur de palladium
Finalement, il est possible d’activer le fluor en condition exclusivement neutre en utilisant des composés donneurs de liaisons hydrogène. Ces méthodes seront détaillées dans la section « liaisons hydrogène » de ce mémoire.
23 Benedetto, E.; Keita, M., Tredwell, M.; Hollingworth, C.; Brown, J. M.; Gouverneur, V. Organometallics
2012, 31, 1408.
Liaisons hydrogène
Une liaison hydrogène, ou pont hydrogène, est une interaction constituée de forces électrostatiques entre un atome d’hydrogène électropositif X‒H (X = N, O, S, F, etc.) et un atome électronégatif Y (Y = N, O, F). Cette interaction vient principalement d’un transfert partiel de charge entre le donneur et l’accepteur résultant en une sorte d’interaction acide-base, mais bien plus faible.
En 2011, l’IUPAC a défini les critères qu’une liaison X‒H…Y doit présenter pour être considérée comme un pont hydrogène. Le critère le plus important est que l’angle de la liaison entre le donneur et l’accepteur doit être le plus près de 180°, plus précisément entre 110 et 180°. Effectivement, une telle orientation facilite le transfert d’une partie de la densité électronique de l’accepteur vers le donneur. Ainsi, plus l’angle de liaison se rapproche de 180°, plus le pont hydrogène est fort. De plus, lors de la formation d’une liaison hydrogène, la longueur de la liaison X‒H augmente, changement qui peut être observé en spectroscopie IR (modification de la fréquence d’élongation νX‒H). Outre la spectroscopie IR, la présence d’une liaison hydrogène est également détectable par spectroscopie RMN, puisque le couplage spin-spin entre Y et X amène une augmentation du déplacement chimique δX-H. Enfin, le dernier critère spécifie que l’énergie thermique du système doit être inférieur à l’énergie de formation de Gibbs, puisqu’il ne s’agirait pas d’une liaison partielle dans le cas contraire.25, 19b
Au début des années 80, Kamlet et Taft ont présenté certains facteurs, appelés paramètres solvatochromiques, qui permettent d’évaluer si un composé est un bon donneur ou accepteur de liaisons hydrogène. Le facteur α montre la capacité d’un composé à transférer partiellement son hydrogène à un accepteur et est principalement basé sur la polarité de celui-ci. Ainsi, plus la liaison X‒H est polaire, plus le facteur α de celle-ci sera grand et plus X‒H sera prompt à être donneur de liaisons hydrogène. Le facteur β, quant
25 (a) Arunan, E.; Desiraju, G. R.; Klein, R. A.; Sadlej, J.; Schneider, S.; Alkorta, I.; Clary, D. C.; Crabtree,
R. H.; Dannenberg, J. J.; Hobza, P.; Kjaergaard, H. G.; Legon, A. C.; Mennucci, B.; Nesbitt, D. J. Pure Appl. Chem. 2011, 83, 1619. (b) Arunan, E.; Desiraju, G. R.; Klein, R. A.; Sadlej, J.; Schneider, S.; Alkorta,
I.; Clary, D. C.; Crabtree, R. H.; Dannenberg, J. J.; Hobza, P.; Kjaergaard, H. G.; Legon, A. C.; Mennucci, B.; Nesbitt, D. J. Pure Appl. Chem. 2011, 83, 1637.
à lui, évalue la capacité de l’atome Y a accepter un proton et est basé sur l’électronégativité de celui-ci. Donc, plus Y est électronégatif, meilleur accepteur de liaison hydrogène il sera.26
La faculté de la liaison C‒F à agir comme accepteur de liaisons hydrogène posa un important débat dans les dernières décennies. D’abord, en analysant les données cristallographiques de la « Cambridge Crystallographic Data Centre », le groupe de Glusker ainsi que Howard, O’Hagan et leurs collaborateurs en sont venus à des conclusions divergentes. En effet, alors que Glusker affirmait que le lien C‒F pouvait agir comme faible accepteur de liaison hydrogène, Howard et O’Hagan ont nié la capacité de celle-ci à agir comme accepteur de liaison hydrogène.27,28 Ensuite, à la fin des années 90, Dunitz et Taylor collaborèrent pour analyser les données cristallographiques de la « Protein Data Bank ». N’ayant trouvé que peu de preuves d’interactions hydrogène avec le lien C‒F, ils publièrent par la suite un célèbre article qu’ils nommèrent « Organic Fluorine Hardly Ever Accepts Hydrogen Bonds ».29 Après ce constat catégorique, il faudra attendre la décennie suivante avant que la question du fluor organique comme accepteur de liaisons hydrogène ne refasse surface.
À la fin des années 2000, de nouvelles études prouvant que la liaison C‒F peut effectuer des interactions hydrogène ont commencé à faire leur apparition. Elles furent revues par Schneider en 2012 qui conclut grâce aux données computationnelles et aux mesures d’équilibre chimique que, en solution ou en phase gazeuse, le fluor organique peut agir comme accepteur de liaisons hydrogène. Comme il s’agit d’une interaction faible, elle ne change pas significativement la façon dont une molécule cristallise, d’où les conclusions écoulées de l’analyse des bases de données cristallographiques.19a Notre groupe de recherche a également, en 2015, revu les nouvelles études montrant les interactions hydrogène que peut effectuer le lien C‒F et est arrivé aux mêmes conclusions que
26 Kamlet, M. J.; Abboud, J.-L. M.; Abraham, M. H.; Taft, R. W. J. Org. Chem. 1983, 48, 2877.
27 (a) Murray-Rust, P.; Stallings, W. C.; Monti, C. T.; Prestone, R. K.; Glusker, J. P. J. Am. Chem. Soc. 1983,
105, 3206. (b) Shimoni, L.; Glusker, J. P. Struct. Chem. 1994, 5, 383
28 Howard, J. A. K.; Hoy, V. J.; O’Hagan, D.; Smith, G. T. Tetrahedron 1996, 52, 12613. 29 Dunitz, J. D; Taylor, R. Chem. Eur. J. 1997, 3, 89.
Schneider, soit que même si elle est de faible énergie, il est difficile de douter de l’existence de la liaison hydrogène C‒F…H‒X à ce jour.19b
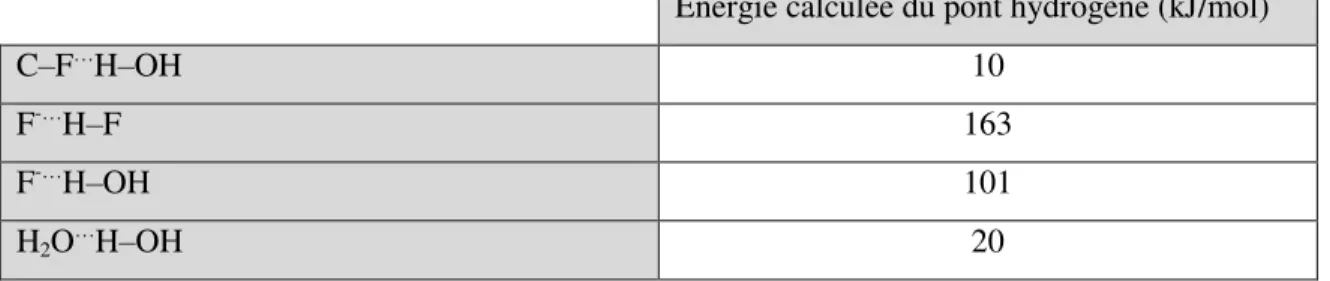
En 2014, Dalvit et Vulpetti se sont intéressés au calcul de la longueur de la liaison hydrogène entre le 4-fluorophénol et différentes molécules organofluorées et non-fluorées (acétophénone). Ils ont ainsi établi une corrélation entre le déplacement chimique en RMN 19F et la longueur du pont hydrogène C‒F…H‒O. Leurs calculs montraient notamment que les composés monofluorés sont de meilleurs accepteurs de liaisons hydrogène que les composés di- et trifluorés. Ils ont également montré que la formation d’une interaction hydrogène entre un composé RCH2F et un donneur était environ 7 kJ/mol moins favorables que celle entre l’acétophénone et un donneur. Étant donné le fort caractère accepteur de liaisons hydrogène de l’acétophénone via le carbonyle de celui-ci, cela montre le potentiel des interactions hydrogène que peuvent former les composés monofluorés. Toutefois, comme mentionné dans l’article, il doit ne pas avoir de compétition avec de meilleurs accepteurs de liaisons hydrogène.30
Même si l’énergie calculée du pont hydrogène C‒F…H‒OH est d’environ 10 kJ/mol, il est intéressant de noter que le fluorure effectue la plus forte interaction hydrogène connue avec HF et effectue une interaction forte avec H2O. À titre comparatif, ces deux interactions sont plus fortes que la liaison hydrogène entre deux molécules d’eau (Tableau i.3).31
30 Dalvit, C.; Invernizzi, C.; Vulpetti, A. Chem. Eur. J. 2014, 20, 11058.
Tableau i.3 : Énergie calculée de quelques liaisons hydrogène
Énergie calculée du pont hydrogène (kJ/mol)
C‒F…H‒OH 10
F-…H‒F 163
F-…H‒OH 101
H2O…H‒OH 20
En observant les différentes méthodes d’activation de liaisons C‒F, par exemple celle avec des acides de Lewis, il est possible de constater que l’interaction C‒F…LA est plutôt faible, mais devient très forte lorsque le fluorure est éjecté. Cela semble également être la force motrice de la réaction. À partir de ce constat, du fait que le lien C‒F peut agir comme faible accepteur de liaisons hydrogène et que le fluorure a la possibilité d’effectuer de fortes interactions hydrogène en solution, notre groupe de recherche s’est demandé s’il était possible de faciliter l’éjection du fluor dans des réactions de substitution en utilisant des composés donneurs de liaisons hydrogène.
Il s’agissait d’un objectif réalisable puisqu’en 2013, le groupe de recherche a montré le premier exemple d’activation de liaisons C‒F benzyliques par des interactions hydrogène avec le solvant réactionnel. Cet exemple, ainsi que les autres méthodes développées par notre groupe de recherche sont détaillées ci-dessous.
Méthodes d’activation de fluor aliphatique par liaisons hydrogène
D’abord, la première méthode présentée par notre groupe de recherche consistait à l’utilisation d’un solvant donneur de liaisons hydrogène pour activer la liaison C‒F benzylique. Lorsqu’un seul solvant donneur (éthanol, isopropanol) était utilisé, la réaction ne fonctionnait pas très bien puisque le fluorure sortant ne semblait pas suffisamment stabilisé. Cependant, lorsqu’un mélange de solvant organique et d’eau en proportion égale était utilisé, des conversions prometteuses étaient obtenues après 4h lorsque l’éthanol ou l’isopropanol étaient utilisés comme solvant. Plusieurs calculs DFT ont été effectués pour mieux comprendre le système. Ceux-ci ont montré qu’une diminution de l’énergie de distorsion de BnF ainsi qu’une interaction F…OH
l’état de transition permettent de stabiliser celui-ci ce qui mène à l’éjection du fluorure. Les analyses DFT ont également montré que la stabilisation du fluor s’effectue via trois liaisons hydrogène avec trois molécules d’eau. Même si le fluor est considéré non-réactif par rapport à la réaction de SN2, la méthode s’est montrée efficace en utilisant différents nucléophiles aminés, oxygénés, soufrés et carbonés et ce, sur des substrats benzyliques, allyliques et sur des cétones α-fluorés. Elle ne fonctionne toutefois pas lorsque des substrats peu activés (alkyles fluorés) ou désactivés (sulfones α-fluorés) sont utilisés (Schéma i.9).32
Schéma i.9 : Activation par une mélange de solvants donneurs de liaisons hydrogène
Par la suite, étant donné que les calculs DFT montraient une stabilisation du fluor sortant par trois molécules d’eau, le groupe de recherche a posé que l’activation serait possible en utilisant une molécule pouvant effectuer à elle seule ces trois ponts hydrogène. En utilisant un triol, soit le 1,1,1-tris(hydroxyméthyl)propane, comme donneur de liaison hydrogène, la réaction de SN2 de différentes amines secondaires sur des fluorures benzyliques a été réalisée (Schéma i.10).33 D’autres analyses par DFT ont été effectuées et ont montré que contrairement à ce qu’il avait pensé, la stabilisation du fluor sortant s’effectuait par deux molécules de triols. Avec deux liaisons hydrogène effectuées par une première molécule de triol et une troisième liaison hydrogène effectuée par une deuxième molécule de triol, l’angle ainsi que la longueur des liaisons C‒F…H‒O permettent des interactions hydrogène optimales. Une version catalytique de la réaction a également été testée. Ainsi, en utilisant 10 mol % de triol et en augmentant le temps réactionnel à 72 h, le groupe était quand même satisfait d’observer une conversion RMN 1H de 77 %. Ceci montrait qu’une activation avec un quantité catalytique de donneur était possible, mais lente. Étant donné la forte interaction qu’effectue le fluorure sortant avec
32 Champagne, P. A.; Pomarole, J.; Thérien, M.-È.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin,
J.-F. Org. Lett. 2013, 15, 2210.
le triol, celui-ci semble se faire rapidement empoisonner une fois que l’activation a lieu d’où le long temps réactionnel.34
Schéma i.10 : Activation par un triol
Ensuite, le groupe s’est demandé s’il était possible, en utilisant un très bon donneur de liaisons hydrogène, d’observer une réactivité de type SN1. En effet, si le donneur de liaison hydrogène est assez fort, il entraînerait l’éjection du fluor ce qui générerait un carbocation sur lequel le nucléophile pourrait attaquer. Les résultats préliminaires montraient qu’en utilisant l’hexafluoroisopropanol (HFIP) comme solvant, le produit obtenu n’est pas celui résultant de la substitution nucléophile avec la morpholine comme attendu, mais bien celui de la réaction de Friedel-Craft du substrat de départ polymérisant avec lui-même. Cela montrait le potentiel de la méthode pour la réaction de SN1 de composés arylés sur les fluorures benzyliques activés. Pour éviter la polymérisation, la proportion de HFIP dans le milieu est diminuée ce qui permet d’obtenir les composés diarylés finaux dans de bons rendements (Schéma i.11). Lors de l’analyse du mécanisme de la réaction, il a été montré que du HF est généré in situ après la première éjection de fluorure. Comme HF est un meilleur donneur de liaisons hydrogène que HFIP, il a été supposé que le système serait autocatalytique par la formation de celui-ci et la période d’induction était estimée à environ 170 minutes. Ainsi, une fois que le HFIP vient activer un premier fluorure benzylique, le HF résultant vient participer à la réaction faisant en sorte qu’elle est de plus en plus rapide à chaque activation. Autre fait intéressant, les tests contrôles de la réaction en utilisant un substrat de départ comportant un fluorure
34 Champagne, P. A.; Drouin, M.; Legault, C. Y.; Audubert, C.; Paquin, J.-F. J. Fluorine Chem. 2015, 17,
benzylique et un chlorure benzylique ont montré que cette méthode de benzylation de Fridel-Craft est sélective aux benzyles fluorés.35
Schéma i.11 : SN1 sur des substrats benzyliques par activation avec HFIP
Finalement, étant donné le système autocatalytique observé pour l’activation par HFIP, le groupe s’est interrogé sur la possibilité d’utiliser une quantité catalytique d’un donneur de liaisons hydrogène pour cette même méthode d’activation. Ainsi, en utilisant un acide donneur comme l’acide trifluoroacétique (TFA), les membres du groupe ont montré qu’une première liaison C‒F est activée et que l’acide accélérait la formation de HF, qui prend le relais pour l’activation. La période d’induction du système était très rapide et était estimée à moins de 10 minutes. Du HFIP est tout de même présent dans le solvant réactionnel puisqu’il semble que celui-ci joue un rôle important dans la stabilisation du système, les rendements isolés étant plus faibles lorsqu’il était absent. Cette réaction est le premier exemple de SN1 sur des benzyles monofluorés en utilisant une quantité catalytique de donneur de ponts hydrogène pour l’activation.36 Plus tard, le groupe a également étendu la méthode à des propargylesmonofluorés, présentant ainsi le premier exemple d’activation de lien C‒F sur ce type de composés (Schéma i.12).37
35 Champagne, P. A.; Benhassine, Y.; Desroches, J.; Paquin, J.-F. Angew. Chem. Int. Ed. 2014, 53, 13835. 36 Hemelaere, R.; Champagne, P. A.; Desroches, J.; Paquin, J.-F. J. Fluorine Chem. 2016, 190, 1. 37 Hamel, J.-D.; Beaudoin, M.; Cloutier, M.; Paquin, J.-F. Synlett 2017, 28, 2823.
Schéma i.12 : réaction de SN1 en utilisant une quantité catalytique de TFA pour
l'activation
Objectifs
Dans ce mémoire, l’accent sera mis sur deux méthodes d’activation de liens C‒F aliphatiques via des interactions hydrogène, soit en utilisant une thiourée comme donneur de liaisons hydrogène et en utilisant un solvant donneur de ponts hydrogène.
D’abord, le projet des thiourées vient faire suite aux méthodes précédemment publiées par le groupe de recherche. En observant celles-ci, on remarque que seule la substitution nucléophile SN2 en utilisant une quantité catalytique de donneurs pour l’activation manque pour compléter le tableau. Comme les thiourées peuvent effectuer au minimum deux liens hydrogène par les deux azotes et que plusieurs exemples dans la littérature montrent qu’elles peuvent catalyser les réactions par des liaisons hydrogène, elles semblaient être des candidates intéressantes pour l’activation de liaisons C‒F.38 L’hypothèse est qu’en utilisant une thiourée comme donneur de liaisons hydrogène, il serait possible d’activer le lien C‒F benzylique en vue d’effectuer la SN2 de divers nucléophiles et que la thiourée pourrait être régénérée à la fin pour effectuer à nouveau l’activation (Schéma i.13). Ces travaux ont récemment été publiés et consistent en un
38 Pour des revues sur l’utilisation de thiourées en synthèse organique, voir : (a) Takemoto, Y. Org. Biomol.
Chem. 2005, 3, 4299. (b) Zhang, Z.; Bao, Z.; Xing, H. Org. Biomol. Chem. 2014, 12, 3151. (c) Gimeno, M.
premier exemple d’activation organocatalysée de liens C‒F sur des substrats benzyliques.39 Ils seront détaillés au chapitre 1 de ce mémoire.
Ensuite, le projet d’activation par un mélange de solvant donneur de liaisons hydrogène est en continuité avec la première méthode publiée par le groupe de recherche. Comme celle-ci représentait le premier exemple d’activation de liens C‒F aliphatiques par des interactions de type ponts hydrogène, l’accent a principalement été mis sur des calculs DFT et des expériences contrôles pour bien comprendre le fonctionnement de la réaction. Les résultats obtenus en utilisant différents nucléophiles et divers substrats de départ étaient toutefois très prometteurs, avec des rendements isolés souvent supérieurs à 70 % pour les substrats de départ benzyliques. Afin de valoriser cette méthode en vue d’applications potentielles en synthèse organique, les conditions réactionnelles ont été davantage optimisées et la réaction a été étendue à d’autres nucléophiles (Schéma i.13). Les résultats obtenus seront décrits au chapitre 2 de ce mémoire.
Schéma i.13 : Projets décrits dans le cadre de ce mémoire
Pour les deux projets, le fluorure benzylique est utilisé comme substrat modèle étant donné sa réactivité favorable vis-à-vis les réactions de substitution nucléophile.
39 Houle, C.; Savoie, P. R.; Davies, C.; Jardel, D.; Champagne, P. A.; Bibal, B.; Paquin, J.-F. Chem. Eur. J.
1
Activation de la liaison C‒F par une thiourée
1.1 Introduction thiourée
L’organocatalyse est un concept de plus en plus présent dans la littérature.40 Il s’inspire des mécanismes enzymatiques présents dans la nature et consiste en l’utilisation de molécules organiques en quantité catalytique, plutôt que des métaux, pour effectuer des transformations chimiques. L’organocatalyse comporte plusieurs avantages au niveau des ressources matérielles tout comme au niveau environnemental et est généralement moins coûteuse que son analogue métallique.40c Les organocatalyseurs fonctionnent également via diverses stratégies. Par exemple, la proline peut permettre d'α-fonctionnaliser les carbonyles via la formation d'une énamine active et nucléophile. Les imidazolidinones, quant à elles, peuvent fonctionnaliser en position β les carbonyles par la formation d'un cation iminium électrophile.40c Une autre méthode courante consiste en l'utilisation de composés donneurs de liaisons hydrogène qui facilite les réactions de substitutions nucléophiles en se complexant avec le substrat par des interactions de type ponts hydrogène. Parmi les composés utilisés pour ce type d'organocatalyse, on retrouve principalement les squaramides, les guanidines, les dérivés d’acides phosphoriques, les urées et les thiourées (Figure 1.1).41,42
Figure 1.1 : Exemples de fonctions chimiques utilisées comme organocatalyseurs
40 (a) Brabas III, C. F. Angew. Chem. Int. Ed. 2008, 47, 42. (b) List, B. Chem. Rev. 2007, 107, 5413. (c)
MacMillan, D. W. C. Nature 2008, 455, 304.
41 Takemoto, Y. Org. Biomol. Chem. 2005, 3, 4299.
Étant donné qu'elles possèdent un grand potentiel donneur de liaisons hydrogène, les dérivés d’urées et de thiourées ont été massivement étudiés.41,43 Par exemple, le groupe de Schreiner a montré l’effet positif amené par l’ajout d’une thiourée à une réaction de Diels-Alder lorsque le substrat de départ est un dérivé d’oxazolidinone. Ici, la thiourée vient se complexer au substrat de départ par deux liaisons hydrogène ce qui favorise la formation d’un des deux diastéréoénantiomères de la réaction et permet même à la réaction d’avoir lieu à température ambiante (Schéma 1.1).44
Schéma 1.1 : Réaction de Diels-Alder catalysée par une thiourée
En utilisant des dérivés chiraux d’urées et de thiourées, il est également possible d’induire de la stéréosélectivité au produit final des réactions. En 2004, le groupe de Nagasawa a développé une version asymétrique de la réaction de Baylis-Hillman en utilisant une bis-thiourée chirale. Selon, le groupe de recherche, la stéréochimie du produit final est déterminée par la coordination du substrat de départ et de l’aldéhyde à la bis-thiourée par des interactions de type liaisons hydrogène (Schéma 1.2). Cependant, cette stéréosélectivité est perdue lorsque des aldéhydes aromatiques sont utilisés, même si ceux-ci permettaient de meilleurs rendements réactionnels.45
43 (a) Kelly, T. R.; Kim, M. K. J. Am. Chem. Soc. 1994, 116, 7072. (b) Schmidtchen, F. P.; Berger, M.
Chem. Soc. 1997, 97, 1609. (c) Linton, B. R.; Goodman, M. S.; Hamilton, A. D. Chem. Eur. J. 2000, 6,
2449.
44 Schreiner, P. R.; Wittkopp, A. Org. Lett. 2002, 4, 217.
Schéma 1.2 : Réaction de Baylis-Hillman asymétrique en utilisant une bis-thiourée chirale
Malgré ces applications intéressantes de l’organocatalyse, il reste beaucoup de chemin à faire avant que celle-ci remplace les catalyseurs métalliques en industrie par exemple. Cependant, étant donné les nombreux avantages que les organocatalyseurs présentent, il reste pertinent de développer de nouvelles méthodes impliquant ceux-ci.
1.2 Résultats préliminaires
Le projet d’activation de liaisons C‒F par liaisons hydrogène avec une thiourée fut débuté par un ancien stagiaire postdoctoral du laboratoire, Dr Paul R. Savoie. Cette section comporte les résultats qu’il avait obtenus, soit le choix de la thiourée, les premiers résultats d’activation en utilisant une quantité catalytique de thiourée ainsi que les problèmes que présentait la méthode à ce moment-là. Mme Clotilde Davies et M. Damien Jardel se sont occupés de la synthèse et la caractérisation des thiourées testées. Pour ma part, j’ai effectué l’optimisation de la version catalytique de la réaction ainsi que l’étendue de la réaction.
1.2.1 Sélection de la thiourée
Étant donné qu’il existe plusieurs possibilités quant aux groupements présents sur les chaînes latérales de la thiourée, il fallait commencer quelque part. Ainsi, notre collaboratrice à l’Université de Bordeaux, la Dre Brigitte Bibal, a fait parvenir une série de thiourées synthétisées par des membres de son laboratoire. La méthode utilisée pour la formation des thiourées consiste en une réaction de SN2 d’amines primaires sur différents isocyanates (Schéma 1.3).
Schéma 1.3 : Méthode de synthèse des thiourées
Pour les premiers tests, 1,1 équivalents de thiourée a été utilisé afin d’investiguer sur le potentiel de celle-ci à activer les liaisons C‒F. Comme substrat de départ, un benzyle monofluoré très activé, soit le 4-(fluorométhyl)-1,1'-biphényl, est utilisé et la morpholine est utilisée comme nucléophile aminé (Schéma 1.4).
Schéma 1.4 : Résultats d'activation du lien C‒F par différentes thiourées
a Conversion RMN 1H calculée à partir du signal des groupements méthylènes benzyliques du produit de
départ et du produit final.
En observant ces résultats, on constate que les thiourées comportant un autre groupement donneur de liaisons hydrogène, soit un alcool, donnaient de meilleures conversions RMN 1H que la thiourée A qui n’en comportait pas. À ce moment, l'hypothèse posée était que ce groupement supplémentaire permettait de stabiliser davantage le fluorure sortant à l’aide d’une troisième liaison hydrogène.
Une augmentation marquée de la conversion est également observée lorsque l’aryle présent sur l’une des chaînes latérales de la thiourée est substitué en position 3,5 par des groupements trifluorométhyles électroattracteurs. Il est possible que ces groupements électroattracteurs viennent rigidifier la conformation de la thiourée par des interactions S‒H favorables, faisant en sorte de diminuer la pénalité entropique observée lors de la complexation avec le substrat.41,42. Il est également possible de poser que l'ajout de ces groupements CF3 vient augmenter la capacité de la thiourée à être donneur de liaisons hydrogène puisqu'ils augmentent l'acidité des amines du motif.
Lorsque la distance entre le groupement alcool et le motif thiourée est augmentée (thiourées C, D et E), on ne note pas de changement important au niveau de la conversion. Lorsqu’un quatrième groupement donneur de ponts hydrogène est ajouté à la chaîne alkyle latérale de la thiourée (F), une conversion très prometteuse de 83 % est obtenue. Cependant, le meilleur résultat obtenu parmi les thiourées testées est celui de la thiourée G. Ici, en ajoutant un groupement ‒CF3 en position α par rapport à l’alcool, on vient augmenter l'acidité de ce dernier ce qui permet d’augmenter son potentiel donneur de liaisons hydrogène. Lorsqu’un deuxième trifluorométhyle est ajouté en α de l’alcool, le potentiel donneur de l’alcool devrait en théorie être augmenté ce qui permettrait une meilleure conversion. Toutefois, lorsqu’on observe la conversion de la thiourée H, on remarque qu’elle est inférieure à celle de la thiourée G, probablement à cause du facteur stérique apporté par l'ajout d'un‒CF3, dont la taille est estimée à celle d’un éthyle.
En somme, l’activation de la liaison C‒F a été observée en utilisant des thiourées en quantité stœchiométrique comme donneur de ponts hydrogène. Pour la suite du projet, soit l’utilisation d’une quantité catalytique de thiourée pour l’activation, deux thiourées ont principalement retenu l’attention, soit la thiourée C et G. La stratégie utilisée consistait en l’utilisation de la thiourée C pour l’optimisation du système puisque celle-ci était facilement synthétisable au laboratoire et avait donné une conversion intéressante de 73 %. Lorsque les conditions optimales seront développées, la thiourée C sera substituée par la thiourée G, puisque celle-ci permettait la meilleure conversion RMN.
1.2.2 Résultats initiaux version catalytique
Maintenant que le choix des thiourées a été effectué, la réaction a été tentée cette fois-ci en utilisant une quantité catalytique du donneur. En utilisant 10 mol % de la thiourée C pour 24 et 48 h, les conversions RMN obtenues étaient respectivement de 8 % et 40 % (Schéma 1.5). Même si la conversion était très faible après 24 h, celle obtenue après deux jours était prometteuse, car elle montrait le potentiel de la méthode à effectuer un cycle catalytique. Tout de même, pour arriver à faire fonctionner la réaction en un temps réactionnel plus court, il était important de comprendre ce qui s’était passé avec la thiourée C pour ces deux temps réactionnels.
Schéma 1.5 : Résultats préliminaires en utilisant une quantité catalytique de thiourée
a Conversion RMN 1H calculée à partir du signal des groupements méthylènes benzyliques du produit de
départ et du produit final.
Dans les 25 dernières années, plusieurs travaux ont été effectués pour la détection d’anions de petite taille, comme le fluorure. D’ailleurs, les premières méthodes pour la détection des anions consistaient à l’utilisation de donneurs de liaisons hydrogène. Sur les molécules utilisées, le site donneur où l’anion venait se coordonner via des ponts hydrogène était voisin à des groupements fluorescents ou sensibles à la réduction. Lorsque l’anion venait se coordonner par interactions hydrogène à cette molécule, cela causait une perturbation électronique au niveau des groupements adjacents qui émettaient alors une réponse détectable. Aujourd’hui, les méthodes les plus courantes de détection d’anion utilisent la fluorescence et la colorimétrie puisque ces méthodes permettent une réponse rapide.46 Dans la littérature, il existe également plusieurs exemples où des
46 Pour des revues sur la détection d’anions, voir : (a) Beer, P. D.; Gale, P. A. Angew. Chem. Int. Ed. 2001,
thiourées sont utilisées pour la détection sélective de fluorure en solution via ces deux méthodes.47
Avec ces exemples en main, il était logique de poser qu’une fois l’activation du fluorure benzylique réalisée, le fluorure sortant était capté par la thiourée comme observé lors de la détection d’anion. Ainsi, cette interaction était peut-être plus forte que celle entre la thiourée et le substrat de départ (Figure 1.2). Sachant également que le fluorure est un meilleur accepteur de liaisons hydrogène que le lien C‒F, il est plausible d’affirmer que la thiourée complexerait prioritairement avec le fluorure ce qui expliquerait la conversion obtenue après 24 h.31 Somme toute, le résultat obtenu après 48 h suggère que cette interaction « fluorure-thiourée » est réversible puisque la thiourée a pu effectuer de l’activation supplémentaire.
Figure 1.2 : Thiourée empoisonnée par le fluorure éjecté
Basé sur ces observations, il a été suggéré qu’en ajoutant un capteur dans la réaction, il serait possible de trapper le fluorure sortant pour empêcher que ce dernier empoisonne la thiourée et ce, en changeant l’équilibre de cette complexation.
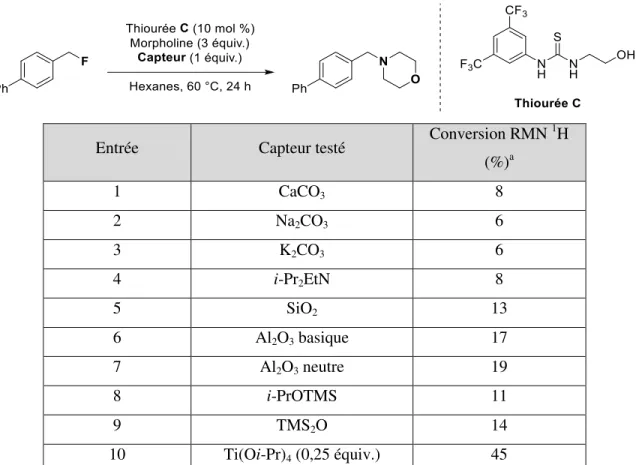
1.2.3 Sélection du capteur
Pour arriver à capter le fluorure sortant, plusieurs composés pouvant effectuer des interactions avec les fluorures ont été testés. Lorsque des bases inorganiques et organiques étaient ajoutées au milieu réactionnel, aucun changement dans la conversion n’était observé (Tableau 1.1, entré 1 à 4). Une modeste augmentation est observée en utilisant des composés adsorbants comme la silice et différentes formes d’alumine
47 Pour des exemples d’articles sur la détection sélective de fluorure par des thiourées, voir : (a)
Chowdhudry, B.; Sinha, S.; Ghosh, P. Eur. J. Org. Chem. 2019, 1008. (b) Dey, S. K.; Hernández, B. G.;
D’Souza, M.; Mhaldar, S. N.; Gobre, V. V.; Dhuri, S. N. ChemistrySelect 2019, 4, 4068. (c) Li, Z.-Y.; Su,
H.-K.; Tong, H.-X.; Yin, Y.; Xiao, T.; Sun, X.-Q.; Jiang, J.; Wang, L. Spectroc. Acta A 2018, 200, 307. (d)
(Tableau 1.1, entrée 5 à 7). Étant donné la forte interaction entre le fluor et le silicium, l’utilisation de composés silylés semblait une avenue intéressante pour trapper le fluorure. Cependant, les résultats étaient semblables à ceux obtenus avec les composés adsorbants (Tableau 1.1, entrée 8 et 9). Enfin, étant donné que le fluor est également connu pour effectuer des liens forts avec les métaux, leur utilisation a été tentée. Puisque l’isopropoxyde de titane (IV) était soluble dans les conditions réactionnelles, il a été testé et une conversion intéressante de 45 % a été obtenue après 24 h en utilisant 0,25 équivalents de celui-ci (Tableau 1.1, entrée 10).
Tableau 1.1 : Résultats obtenus lors de la sélection du capteur
Entrée Capteur testé Conversion RMN
1H (%)a 1 CaCO3 8 2 Na2CO3 6 3 K2CO3 6 4 i-Pr2EtN 8 5 SiO2 13 6 Al2O3 basique 17 7 Al2O3 neutre 19 8 i-PrOTMS 11 9 TMS2O 14 10 Ti(Oi-Pr)4 (0,25 équiv.) 45
a Conversion RMN 1H calculée à partir du signal des groupements méthylènes benzyliques du produit de
départ et du produit final.
Il est difficile de tirer des conclusions à propos de la sélection du capteur. Il est vrai que la liaison Ti‒F est forte avec une énergie d’environ 569 kJ/mol, mais la liaison Si‒F est