Nouvelle méthode d’activation du lien C-F promue par
des donneurs de liaisons hydrogènes
Mémoire
Yasmine Benhassine
Maîtrise en chimie
Maître ès sciences (M.Sc.)
Québec, Canada
©Yasmine Benhassine, 2016
Nouvelle méthode d’activation du lien C-F promue par
des donneurs de liaisons hydrogènes
Mémoire
Yasmine Benhassine
Sous la direction de :
Résumé
La liaison C-F est la plus forte liaison simple que peut faire le carbone, elle est aussi particulièrement plus forte que toutes les autres liaisons carbone-halogène (enthalpie de dissociation à 298 K: CH3-F 480,7 kJ/mol; CH3-Cl 349,9 kJ/mol, etc.). Malgré la force de
cette liaison, de nombreuses méthodes sont apparues dans les dernières années pour l’activer dans les composés aromatiques, vinyliques et aliphatiques. Typiquement, ces méthodes nécessitent des conditions fortement acides ou l'utilisation de métaux de transition.
Nous avons développé une méthode d’activation de liaisons C-F par l’eau pour réaliser des réactions de substitution nucléophile de type SN2. La réactivité accrue du lien carbone-fluor
est due aux liaisons hydrogène entre l'eau et l'atome de fluor, qui agit comme accepteur. Suite à ces travaux, nous avons étudié la réactivité des fluorures benzyliques en présence de donneurs de liaisons hydrogène plus forts que l’eau, tel que le 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP). Les résultats obtenus démontrent une réactivité inédite de type Friedel-Crafts (SN1). Une optimisation de la réaction a été effectuée.L'étendue de la
transformation a été étudiée et les résultats obtenus nous ont permis de comprendre la réactivité du système en fonction des propriétés électroniques des fluorures de benzyle et des nucléophiles. Tous ces travaux ainsi que des études mécanistiques préliminaires seront présentés.
Abstract
The carbon-fluorine bond is the strongest single bond that a carbon can have, it is also particularly stronger than any other carbon-halogen bonds (dissociation enthalpy at 298 K: CH3-F 480,7 kJ/mol; CH3-Cl 349,9 kJ/mol.). Despite the strength of this connection,
numerous methods have emerged in recent years to turn it into aromatic and aliphatic vinyl compounds. Typically, these methods require strongly acidic conditions or the use of transition metals.
We developed a C-F activation method with water to produce nucleophilic substitution reactions type SN2. The reactivity of carbon-fluorine bond is due to the hydrogen bonds
between water and the fluorine atom, which acts as an acceptor.
Following this work, we investigated the reactivity of benzyl fluoride in the presence of stronger hydrogen bond donor than water, such as 1,1,1,3,3,3- hexafluoro-2-propanol ( HFIP) The results show a Friedel-Crafts type unprecedented reactivity (SN1). An
optimization of the reaction was carried out. The extent of the transformation has been studied and the results obtained have allowed us to understand the reactivity of the system based on electronic properties of benzyl fluorides and nucleophilic. All this work and preliminary mechanistic studies will be presented
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... vii
Liste des schémas ... viii
Listes des Figures ... x
Liste des abréviations ... xi
Remerciements ... xiii
1. Introduction ... 1
1.1 L'histoire du fluor: ... 1
1.2 Les propriétés de l’atome de fluor : ... 2
1.3 La liaison carbone-fluor (C-F) : ... 2
1.4 Activation de la liaison C-F aliphatique dans les composés monofluorés ... 3
1.5 Résultats précédents ... 10
2. La réaction de Friedel-Crafts (SN1) ... 14
2.1 Alkylation de Friedel-Crafts ... 14
2.2 Benzylation de Friedel-Crafts : ... 16
2.3 Alkylation de Friedel-Crafts avec des fluorures d'alkyles : ... 18
2.4 Objectif du projet ... 21
3. Synthèse de fluorures de benzyles ... 22
3.1. Intérêt des fluorures de benzyles ... 22
3.2. Synthèses des substrats ... 22
3.2.1. Par fluoration de bromures de benzyles : ... 22
3.3 Optimisation des conditions de la réaction ... 23
3.3.1 Optimisation préliminaire: cas du 1,4-diméthoxybenzène ... 23
3.3.2 Optimisation en présence de p-xylène ... 26
3.4 Étendue de la réaction de substitution nucléophile ... 28
3.6 Effet du degré de substitution par le fluor ... 47
3.7 Comparaison de différents groupes partants ... 49
3.7.1 Hypothèse mécanistique : ... 51
Liste des tableaux
Tableau 1. Valeurs de pKa et α de trois solvants ... 12
Tableau 2. Optimisation du nombre équivalents avec le 1,4-diméthoxybenzène... 25
Tableau 3. Optimisation du nombre d’équivalents avec le p-xylène ... 26
Tableau 4. Optimisation du solvant dans la réaction de substitution nucléophile ... 27
Tableau 5. Optimisation de la réaction avec le chloroforme ... 28
Tableau 6. Étendue de la réaction de Friedel-Crafts avec des groupements méthyles ... 31
Tableau 7. Étendue de la réaction de Friedel-Crafts avec des nucléophiles non substitués. 33 Tableau 8. Étendue de la réaction de Friedel-Crafts avec des hétérocycles aromatiques .... 35
Tableau 9. Étendue de la réaction de Friedel-Crafts avec des substituants halogénés ... 37
Tableau 10. Étendue de la réaction de Friedel-Crafts avec des substituants carbonylés ... 39
Tableau 11. Étendu de la réaction de substitution nucléophile avec des fluorures de chlorobenzyles et de bromobenzyles ... 42
Tableau 12. Étendu de la réaction de substitution nucléophile avec des fluorures benzyliques contenant des groupements donneur ... 44
Tableau 13. Étendu de la réaction de substitution nucléophile avec des fluorures benzyliques secondaire ... 47
Tableau 14. Étendu de la réaction du substrat difluoré ... 48
Tableau 15. Étendu de la réaction du substrat trifluoré ... 49
Liste des schémas
Schéma 1. Activation d’un monofluorure allylique par un catalyseur de Pd ... 4
Schéma 2. Substitution nucléophile de l’atome de fluor avec un acide de Brønsted ... 4
Schéma 3. Activation de la liaison C-F par un acide de Lewis ... 5
Schéma 4. Réaction de glycosylation réalisée à l'aide d'acides forts de Lewis ... 5
Schéma 5. Activation de la liaison C-F Par l’utilisation de cations (Silylium) ... 6
Schéma 6. Activation de la liaison C-F en milieu basique ... 6
Schéma 7. Réaction développée par le groupe de Leckta qui démontre la capacité du fluor a accepter des liaisons hydrogène. ... 9
Schéma 8. Exemple développé par le groupe Linclau sur les alcools qui démontre la capacité du fluor a accepter des liaisons hydrogène. ... 9
Schéma 9. Activation de la liaison C-F en milieu neutre ... 10
Schéma 10. Mécanisme et structure du produit proposés pour la formation du polymère insoluble lors de la mise en solution du fluorure benzylique dans le HFIP. ... 13
Schéma 11. Réaction de solvolyse ... 13
Schéma 12. Alkylation de Friedel-Crafts ... 14
Schéma 13. Schéma général de la benzylation de Friedel-Crafts ... 16
Schéma 14. Benzylation de Friedel-Crafts en présence de chlorure d'aluminium (AlCl3)... 16
Schéma 15. Benzylation de Friedel-Crafts en présence de trifluorure de bore (BF3) ... 17
Schéma 16. Benzylation de Friedel-en présence de trichlorure de fer (FeCl3) ... 17
Schéma 17. Benzylation de Friedel-Crafts en présence de métaux de transition ... 18
Schéma 18. Benzylation de Friedel-Crafts en présence d'acide de Brønsted. ... 18
Schéma 19. Activations de fluorures d'alkyle avec des acides de Lewis ... 19
Schéma 20. Activation de fluorure d'alkyle en présence de cation silylium catalytique ... 19
Schéma 21. Activation de fluorure d'alkyle par un acide fort de Brønsted ... 19
Schéma 22. Optimisation des conditions de la réaction ... 21
Schéma 23. Synthèse du précurseur bromé ... 22
Schéma 24. Étape finale de fluoration ... 23
Schéma 25. Réaction de déoxofluoration ... 23
Schéma 27. Le fluorure de 4-phénylbenzyle dans les conditions optimales ... 40
Schéma 28. Le produit de dialkylation ... 43
Schéma 29. Mécanisme de polymérisation ... 45
Schéma 30. Réaction de Friedel-Crafts avec le XtalFluor-E ... 45
Schéma 31. Synthèse du substrat difluoré. ... 48
Listes des Figures
Figure 1. Différents composés organofluorés ... 3 Figure 2. Calculs DFT de l'état de transition de la substitution nucléophile. ... 11 Figure 3. Classement des substituants en fonction de leur caractère donneur et attracteur. . 15 Figure 4. Nombre de publications sur la Friedel-Crafts de 1928 à 2007. ... 15 Figure 5. Réaction test avec différents nucléophiles substitué avec des groupements
méthoxy ... 29 Figure 6. Diagramme d’énergie de dissociation ... 51
Liste des abréviations
DAST trifluorure de diéthylamino soufre DCE 1,2-dichloroéthane
DFT Density Functional Theory
DLH donneur de liaisons hydrogène HFIP 1,1,1,3,3,3-hexafluoropropan-2-ol RMN résonance magnétique nucléaire
SN1 substitution nucléophile unimoléculaire
SN2 substitution nucléophile bimoléculaire
Ph phényle
TBAF fluorure de tétra-n-butylammonium
t-Bu tert-butyle
TFE 2,2,2-trifluoroéthanol THF tétrahydrofurane
“Plus grand est l'obstacle, et plus grande est la gloire de le surmonter.” Molière
Remerciements
Les études graduées ont été pour moi une expérience enrichissante, elles m’ont permis aussi de me réaliser et de me remettre à niveau. Ce parcours a été formateur et très agréable avec la présence de personnes au sein du groupe de recherche et en dehors qui m'ont permis d'évoluer, d'apprendre et de cheminer sur la bonne voie.
Je tiens en premier à remercier mon directeur de recherche Jean-François Paquin pour l'énorme chance qu'il m'a donnée, ainsi que pour son aide précieuse et sa disponibilité tout le long de ma maitrise. Il était un professeur et un exemple exceptionnel de gentillesse et d'humilité et a su me diriger dans un système qui était nouveau pour moi. Je ne vous remercierai jamais assez de m'avoir donné cette chance et de m'avoir fait confiance.
Un merci spécial à mon ami et collègue Pier Alexandre Champagne qui répondait présent à toutes les fois que je l'ai sollicité, et on sait que le nombre est incalculable. Tu as été pour moi un mentor, sans toi je n'aurai pas eu la même vision que j'ai de mon expérience à présent. Merci de m'avoir appuyé dans les moments heureux et difficiles. En plus d'être un mentor, tu as su être un ami fidèle avec qui j'ai pu partager beaucoup d’aventures.
On n'oublie pas le groupe Paquito composé principalement d'Elsa, Myriam et Justine avec qui j'ai partagé des moments de folies en musique. Merci les filles!!
J’aimerais également remercier TOUS mes collègues de travail anciens et actuels du groupe Paquin que j’ai eu la chance de côtoyer au cours de ces deux dernières années et qui m’ont également beaucoup aidé. Je ne pourrai jamais oublier tous les bons moments que nous avons passés ensemble. D'autres personnes importantes dans ma vie ont contribué de très près malgré la distance qui nous séparait, mes parents Zora et Lazhar que je remercie, car ils ont toujours cru en moi et m'ont encouragé dans toutes mes décisions afin que je puisse réaliser mes rêves. Merci maman pour ton soutien, je n'aurais rien pu faire sans ta présence, tu es et tu seras toujours un modèle de perfection pour moi. Merci à ma sœur et à mon frère Besma et Borhane avec qui j'ai partagé un merveilleux bout de vie.
Également je tiens à remercier ma tante Nabila pour son accompagnement dans mon processus d'adaptation au Québec. Merci à ma meilleure amie Manel qui a su me changer les idées chaque fois que j'en avais besoin, Skype et toi m'aviez comblé.
Finalement, ma vie ne serait pas ce qu’elle est sans la présence de mon partenaire de vie Amine qui a été là jour après jour à m’appuyer dans toutes mes décisions, à me réconforter, à me faire rire et surtout à avoir cru en moi. Tu as été d’une patience remarquable face à la boule de nerf que je suis, merci d’être là dans ma vie et de continuer de l'être malgré la distance qui nous sépare.
Ps : Un petit coucou à Olivier Mahé qui m’a aidé à revoir l’ensemble du mémoire, surtout pour les fautes de synthaxe et les formulations de phrases.
1. Introduction
1.1 L'histoire du fluor:
C'est en 1886, à la faculté de Pharmacie de Paris, qu'Henri Moissan a réussi à isoler le fluor élémentaire pour la toute première fois. Cette découverte lui permit d'obtenir le prix Nobel en 1906. Malgré la découverte de cet élément chimique, la chimie des composés organofluorés fut très lente à démarrer. A cette époque, Henri Moissan consacra seulement 26 de ses 303 pages de son livre aux dérivés organiques, principalement aux fluorures d'alkyles. La chimie des dérivés fluorés organiques est longtemps restée une curiosité en laboratoire.1
La chimie du fluor a joué un rôle significatif dans divers développements technologiques au cours des 80 dernières années. Par exemple, l'une des premières utilités de cet élément fut la fabrication des gaz fréons utilisés pour la réfrigération. Par la suite, la chimie du fluor a connu un essor important en rapport avec le projet Manhattan pour la réalisation d'armes nucléaires. Afin de mettre en marche ce projet, l'enrichissement isotopique de l'uranium était requis. Les chercheurs de l'époque transformèrent l'uranium sous forme gazeuse en hexafluorure d'uranium. Pour ce faire, ils ont eu recours à une grande quantité d'acide fluorhydrique et de fluor élémentaire pour produire ce gaz.2
De plus en plus populaire, la chimie du fluor a participé au développement technologique dans le domaine des polymères ainsi que dans les secteurs pharmaceutique et agrochimique. En effet, c'est durant les années 1950 que les premières grandes industries comme Dupont de Nemours ont exploité la chimie du fluor pour le développement de matériaux très prometteurs comme le Téflon™. De plus, les découvertes des anesthésiques généraux fluorés et des propriétés anti tumorales de composés naturels fluorés permirent de donner naissance à un tout nouveau domaine : la chimie médicinale des composés organofluorés.3
1Tressaud, A. Angew. Chem. Int. Ed. 2006, 45, 6792.
2 Hiyama, T. In organofluorine compounds; chemistry and applications; Yamamoto, H., Ed.; Springer: New
York, 2000.
1.2 Les propriétés de l’atome de fluor :
Le fluor est un élément chimique de symbole F et de numéro atomique 9. Il s'agit du premier élément de la famille des halogènes, de masse atomique 19. C’est l’élément chimique le plus réactif, et il possède des propriétés fortes intéressantes qui sont :
Un rayon de Van der Waals de 1,47 Å. Une taille entre celle de l'atome d'hydrogène, qui est de 1,20 Å, et celle de l'atome d'oxygène, qui est de 1,52 Å.4 De ce fait, la substitution
d'un atome d'hydrogène ou d'oxygène par un atome de fluor ne provoque généralement que de faibles perturbations stériques. Cependant, il en va tout autrement lors du remplacement d'un groupement méthylène (CH2) par un CF2 et même d'un CH3 par un CF3. En effet, ces
groupements fluorés sont beaucoup plus volumineux que leur homologue non-fluoré.1,5
Avec sa configuration ls2 2s22p5, le fluor est l'élément le plus électronégatif du tableau
périodique (3,98 sur l'échelle de Pauling),6 ce qui lui donne un puissant effet inductif
élèctro-attracteur. Sa petite taille et sa forte électronégativité font du fluor un atome très peu polarisable. Aussi, l'introduction sélective d'un atome de fluor peut souvent changer l'activité biologique d'un composé en modifiant certaines propriétés physico-chimiques comme son pKa, sa lipophile, sa conformation ainsi que son aptitude à créer des interactions électrostatiques. Les propriétés métaboliques vont parfois être également modifiées puisque la présence d'un atome de fluor peut dans certains cas ralentir le métabolisme oxydatif, protéger un principe actif du métabolisme d'hydrolyse ou encore éviter une racémisation in vivo.
1.3 La liaison carbone-fluor (C-F) :
La liaison carbone-fluor est la liaison simple la plus forte que peut former le carbone (486 kJ/mol)avec tout autre atome. À titre comparatif, l'énergie de la liaison C-H est de 414 kJ/mol et celle du lien C-Cl est de 461 kcal/mol.2De plus, cette force de liaison s'accentue
directement avec le nombre d'atomes de fluor portés par le carbone, contrairement aux autres halogènes.
4 Hiyama, T. Organofluorine Compounds; Chemistry and Applications, Yamamoto, H. (Éd.), Springer, 2000. 5 Bott, G.; Field, L. D.; Sternhell, S. J. Am. Chem. Soc. 1980, 102, 3618.
Ces facteurs réunis font que l'ion fluorure soit un très mauvais groupe partant en chimie organique malgré le fait que la polarisation de la liaison C-F aurait suggéré une attaque nucléophile facile sur le carbone portant une grande charge partielle positive. Récemment, la liaison C-F a été au centre des intérêts de plusieurs groupes de recherches visant le développement de nouvelles méthodes d’activation de cette liaison qui est difficile à faire réagir.
On peut classer les composés organofluorés en trois types, qui décrivent l’atome de carbone auquel l’atome de fluor est lié (Figure 1). Ainsi, le fluor peut être porté par un carbone hybridé sp (fluorure alcynique), sp2 (fluorure aromatique ou vinylique) ou sp3 (fluorure
aliphatique) et les méthodes pour activer chacun de ces types sont spécifiques.3 On peut
aussi avoir plus d’un fluor sur le même carbone, donnant lieu à des composés CF2 ou CF3
qui ont une réactivité bien différente de leurs analogues monofluorés. Au cours de mon projet de maitrise, je me suis limité à l’étude d’activation des monofluorés aliphatiques tout particulièrement à l’étude de la réactivité des fluorures benzyliques.
Figure 1. Différents composés organofluorés
1.4 Activation de la liaison C-F aliphatique dans les composés monofluorés
Il existe de nombreuses méthodes pour substituer les fluorures d'alkyle, dans lesquelles des conditions acides, basiques, ou l'utilisation de métaux de transition sont mis en œuvre. En voici quelques exemples.
La réaction ci-dessous a été effectuée par le groupe de Gouverneur qui a mis au point un protocole pour activer une liaison C-F à l’aide d’un métal de transition (Schéma 1).
Notamment, cette transformation nécessite un catalyseur de palladium et permet le remplacement du fluor par un malonate.7
Schéma 1. Activation d’un monofluorure allylique par un catalyseur de Pd
Ensuite, l’activation de la liaison C-F peut être effectuée à l’aide d'acides forts de Brønsted. Un exemple très intéressant de ce dernier a été publié en 1990 par le groupe de Barrio. En effet, il y est démontré que lorsqu’on substitue indépendamment le fluorure, chlorure et bromure de cyclohexyle par de l’acide iodhydrique HI (57% dans l’eau), la conversion du fluorure secondaire atteint 64% alors que celle du chlorure ou du bromure est au moins 20% plus faible pour un même temps de réaction.8
Cet exemple confirme donc bien qu’en milieu acide, on assiste à une activation particulière de la liaison C-F par rapport aux autres halogènes (Schéma 2).
Schéma 2. Substitution nucléophile de l’atome de fluor avec un acide de Brønsted
Le même genre de réactivité est obtenu en présence d'acides forts de Lewis. Cette chimie est beaucoup plus développée que les autres exemples, car la capacité de base de Lewis du fluor est bien connue et a été exploitée à maintes reprises. Un de ces exemples provient du groupe d'Olah qui a utilisé le trifluorure de bore afin d’activer la liaison C-F et permettre
7 Hazari, A.; Gouverneur, V.; Brown, J. M. Angew. Chem. Int. Ed. 2009, 48, 1296.
l’attaque du nucléophile, dans ce cas le benzène, et ainsi former le produit désiré à 94%
(Schéma 3).9
Schéma 3. Activation de la liaison C-F par un acide de Lewis
Un autre exemple très intéressant provient de la chimie des sucres, où l'utilisation d'un fluorure de glycosyle comme donneur dans la réaction de glycosylation permet d'obtenir le disaccharide avec de meilleurs ratios α/β qu'en utilisant les autres halogénures de glycosyle.10 (Schéma 4).
Schéma 4. Réaction de glycosylation réalisée à l'aide d'acides forts de Lewis
Une autre méthode pour l’activation de composés aliphatiques fluorés est l’utilisation de cations. Dans la dernière décennie, une nouvelle variante de ce procédé a été décrite dans la littérature et tire profit de la forte affinité des cations silylium pour les fluorures. En effet, le groupe d'Ozerov a démontré en 2005, puis en 2008, la possibilité d’utiliser un système catalytique en cations silylium pour abstraire tous les fluors d’un perfluoroalcane et réaliser une réaction complète d’hydrodéfluoration.11,12 (Schéma 5)
9 Olah, G. A.; Kuhn, S. J. Org. Chem. 1964, 29, 2317.
10 Mukaiyama, T.; Murai, Y.; Shoda, S. Chem. Lett. 1981, 3, 431.
11 Scott, V. J. ; Çelenligil-Çetin, R.; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 2852. 12 Douvris, C.; Ozerov, O. V. Science 2008, 321, 1188.
Schéma 5. Activation de la liaison C-F Par l’utilisation de cations (Silylium)
On sait également que les fluorures d’alkyles sont considérés comme non réactifs dans des conditions neutres ou basiques et cette faible réactivité se traduit par le petit nombre de publications scientifiques décrivant une réaction de substitution nucléophile sur ces substrats. Ainsi, le groupe de Tani et Otsuka a publié en 1982, l'utilisation d'un phosphure de lithium, espèce fortement nucléophile et basique, pour la substitution de la liaison C-F.
(Schéma 6)
Schéma 6. Activation de la liaison C-F en milieu basique
Mon collègue Pier Alexandre Champagne se basant sur des résultats de précédents projets du groupe Paquin, a étudié lors de ses études doctorales l'activation de liaisons C-F par un donneur de liaison hydrogène comme l'eau, et ainsi développé des conditions de substitution nucléophile de fluorures aliphatiques en milieu faiblement basique. Les résultats obtenus très concluants ont permis de démontrer que la liaison C-F (notamment benzylique) peut être activée par différent donneurs de liaisons hydrogène et être substituée.
Qu’est-ce qu’une liaison hydrogène?
X-H donneur de liaison hydrogène (X = N, O, F, etc.)
La liaison hydrogène (HB) ou pont hydrogène est une liaison de faible intensité environ vingt fois plus faible qu'une liaison covalente. Elle implique un donneur de liaison hydrogène (X-H) et un accepteur comme l’azote, l’oxygène ou le fluor, au sein d'une même molécule ou entre plusieurs molécules.
La donation de liaisons hydrogène est un phénomène très proche de l’acidité. C’est une donation de proton ou une interaction hydrogène ayant lieu entre l’accepteur et le donneur de liaison hydrogène. Il faut savoir que si cette interaction se renforce jusqu'au transfert total de l’hydrogène du donneur à l’accepteur on parle de comportement acide. Dans ce cas, il ne s’agit pas d'une donation de liaison hydrogène.13
La question qui se pose dans le cas de nos réactions est : le fluor est-il un accepteur de liaison hydrogène?
Pendant un certain temps, les avis étaient partagés. En effet, si on regarde les énergies d’interaction entre un hydrogène et un fluor calculé par le groupe Emsley,14 on
remarque que dans le cas du fluorure, l’énergie de l’interaction est très forte (155 kJ/mol), alors que dans le cas où le fluor est lié à un atome de carbone l’interaction est de seulement 10 kJ/mol.
Dans The Nature of the Chemical Bond,15 Pauling a démontré que la capacité du
lien hydrogène à être accepté est généralement lié à l'électronégativité de l'accepteur, comme le prouve la forte liaison hydrogène observée pour les fluorures inorganiques. Cependant, il en va tout autrement dans le cas des fluors organiques (lien C-F) pour lesquels aucun exemple n'a permis de prouver cette affirmation.
Ces premières observations par Pauling ont ouvert un débat dans le monde de la chimie sur la capacité du fluor organique à agir comme accepteur de liaison hydrogène. D'abord en 1983 et ensuite en 1994, Glusker et ses collaborateurs ont cherché dans les structures du
13 Pihko, P. M. Hydrogen bonding in Organic Synthesis, Wiley-VCH, Weinheim, 2009. 14 Emsley, J. Chem. Soc. Rev. 1980, 9, 91.
Cambridge Crystallographic Database afin de déterminer les interactions C-F……H. Suite à
cette recherche, les chercheurs ont assuré que la liaison C-F peut agir comme accepteur faible.16 En 1996, Howard, O'Hagan et ses collaborateurs ont réalisé une autre analyse
toujours à partir des structures provenant du Cambridge Crystallographic Database et sont arrivés à une vue légèrement différente du problème en concluant que le fluor est un faible accepteur de liaison hydrogène.17 Dunitz et Taylor, en 1997, ont dirigé une analyse
crystallographique qui a inclus, en plus du Cambridge Crystallographic Database, The
protein Data Bank, ce qui leur a valu une publication très notoire du nom de Organic Fluorine Hardly Ever Accepts Hydrogen Bonds.18 Après cela, peu de publications portant
sur ce sujet furent publiées.
Ce n'est qu’en 2011 que le groupe de Leckta a publié un papier démontrant que le fluor est en effet un accepteur de liaison hydrogène.19 Pour cela, la basicité de trois molécules
différentes a été comparée. Deux de ces molécules portaient un atome de fluor et on s’attendait à ce qu'elles soient moins basique étant donné le caractère électronégatif de ce dernier. Contre toute attente, il a été observé qu'une des molécules fluorées est plus basique que le dérivé non-fluoré. Dans cette molécule, après ajout d'un acide de Brønsted, on observe la formation d’une liaison hydrogène avec le fluor qui stabilise la charge positive. La forme acide est donc stabilisée d’où la basicité supérieure de cette molécule.
Cet exemple vient nous confirmer la capacité de l’atome de fluor à accepter des liaisons hydrogène (Schéma 7).
16 (a) Murray-Rust, P.; Stallings, W. C.; Monti, C. T.; Prestone, R. K.;Glusker, J. P. J. Am. Chem. Soc. 1983,
105, 3206. (b) Shimoni, L.; Glusker, J. P. Struct. Chem. 1994, 5, 383.
17 Howard, J. A. K.; Hoy, V. J.; O’Hagan, D.; Smith, G. T. Tetrahedron 1996, 52, 12613. 18 Dunitz, J. D.; Taylor, R. Chem. Eur. J. 1997, 3, 89.
19 Scerba, M. T.; Leavitt, C. M.; Diener, M. E.; DeBlase, A. F.; Guasco, T. L.; Siegler, M. A.; Bair, N.;
Schéma 7. Réaction développée par le groupe de Leckta qui démontre la capacité du fluor a accepter des liaisons hydrogène.
Un autre exemple développé par le groupe de Linclau vient supporter les résultats émis par le groupe de Leckta en comparant la capacité de fonctions alcools portées par deux molécules fluorées, à donner une liaison hydrogène à un accepteur externe (Schéma 8).20 Il
est montré que le dérivé ne possédant qu'un atome de fluor est meilleur donneur de liaison hydrogène. Ceci s’explique par la formation d’une liaison hydrogène intramoléculaire entre la fonction alcool et le fluor en position alpha dans la molécule difluorée. La conformation comportant une liaison hydrogène avec le fluor est présente majoritairement à 97,8% ce qui empêche la molécule de former une liaison hydrogène avec une autre molécule. Ces deux exemples viennent confirmer que le fluor est bien un accepteur de liaison hydrogène.
Schéma 8. Exemple développé par le groupe Linclau sur les alcools qui démontre la capacité du fluor a accepter des liaisons hydrogène.
Depuis cette période, de nombreuses autres études ont été publiées et la majorité d’entre elles supportent l’idée qu’un lien C–F peut réellement être un accepteur de liaisons hydrogène. Notons que la revue publiée par Schneider en 2012,21 puis par mes deux
collegues Pier-Alexandre Champagne et Justine Desroches en 2015,22 arrivent
pratiquement aux mêmes conclusions. Les conclusions de Schneider sont que les mesures en solution ou en phase gazeuse ont habituellement donné des résultats constants et positifs,
20 Graton, J.; Wang, Z.; Brossard, A.-M.; Gonçalves Monteiro, D.; Le Questel, J.-Y.; Linclau, B. Angew.
Chem. Int. Ed. 2012, 51, 6176.
21 Schneider, H.-J. Chem. Sci. 2012, 3, 1381.
en faveur de la liaison hydrogène avec le fluor. Ces résultats sont corroborés par les études computationnelles, ainsi que les mesures d’équilibres chimiques. Il souligne également que contrairement à toutes ces preuves, les analyses de bases de données cristallographiques n’ont jamais donné de résultats concluants, probablement car l’interaction C–F···H est trop faible et est donc supplantée par la somme de toutes les autres interactions non-covalentes lorsqu’une molécule cristallise. Selon le groupe, il est très difficile de nos jours de douter de l’existence des liaisons hydrogène avec le fluor organique comme accepteur. Cependant, comme c’est une interaction faible, il est important de la caractériser à l’aide d’un maximum de méthodes, en évitant de simplement remarquer une courte distance X(H)···F et de l’attribuer à une interaction attractive. Il a été donc évalué que plusieurs exemples sont rapportés comme une liaison hydrogène alors que les preuves ne supportent pas une telle évaluation.
1.5 Résultats précédents
Dans cette optique, mon collègue Pier Alexandre Champagne a voulu explorer la capacité de l'eau à activer la liaison C-F.
Le (Schéma 9) décrit cette réaction entre la morpholine et un fluorure benzylique, en présence d'eau comme donneur de liaison hydrogène pour l'activation de l’atome du fluor. Le produit désiré est formé à 96% avec des conditions très simples.
Schéma 9. Activation de la liaison C-F en milieu neutre
Ce résultat semble indiquer que la réactivité observée provient d'une liaison hydrogène entre l'eau et le fluorure aliphatique, ce qui affaiblirait la liaison C-F et permettrait donc sa substitution par un nucléophile.
Pour appuyer notre hypothèse, le professeur Legault de l'Université de Sherbrooke a effectué des calculs DFT. La DFT est une méthode de calcul quantique permettant l'étude
de la structure électronique, dans ce cas elle a été effectué afin d’étudier l’état de transition de la substitution nucléophile sans eau et en présence d’eau. (Figure 2)
Figure 2. Calculs DFT de l'état de transition de la substitution nucléophile.23
Il a été remarqué que sans eau, l’énergie de l’état de transition est de 22,4 kcal/mol, tandis qu'en présence de trois équivalents d’eau, l’énergie de l’état de transition est de 8,8 kcal/mol. Nous pouvons en conclure qu'effectivement, l'eau participe, à travers la donnation de liaison hydrogène à l'atome de fluor, à affaiblir la liaison C-F et et à faciliter la réaction SN2.
Ces résultats ont permis de cerner le rôle de l'eau en tant qu'agent d'activation lors de la réaction SN2 de différents fluorures d'alkyles activés.24 Ils ont permis également de
développer un système de solvants i-PrOH : H2O (1 : 1) qui permet de réaliser des réactions
SN2 sur différents motifs fluorés à l'aide d'une vaste gamme de nucléophile et ce dans des
conditions presque neutres (faiblement basiques). Une telle réactivité n'avait jamais été décrite auparavant dans la littérature.
Dans le but de vérifier cette hypothèse, l'utilisation de donneurs de liaisons hydrogénes meilleurs que l'eau semblait toute désignée. Nous avons alors remplacé l'eau par le 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) (facteur α de donations de liaisons hydrogène: 1,96 vs 1,17 pour H2O)25 et le 2,2,2-trifluoroéthanol (TFE) (α : 1,51) dans notre réaction
modèle (Tableau 1).
23 Champagne, P. A.; Pomarole, J.; Thérien, M.-E.; Benhassine, Y.; Beaulieu, S.; Legault, C. Y.; Paquin,
J.-F. Org. Lett. 2013, 15, 2213.
24 Schadt, F. L.; Bentley, T. W.; Schleyer, P. V. R. J. Am. Chem. Soc. 1976, 98, 7667.
La valeur alpha permet de comparer la capacité a donner des liaisons hydrogène. Cette échelle a été développée en 1989 par Kamlet et Taft25.
Tableau 1. Valeurs de pKa et α de trois solvants
Solvant pKa Α
H2O 15,74 1,17
TFE 12,5 1,51
HFIP 9,3 1,96
Dans ces conditions, la réaction avec le HFIP a fournit un résultat très innatendu. En effet, avant même que l'amine ne soit ajoutée à la solution du fluorure benzylique dans ce solvant, une réaction très rapide était observée, menant à la formation presque instantanée d'un solide blanc. Étant insoluble dans la plupart des solvants habituels, nous avons supposé que ce solide avait probablement une structure polymérique.
Malgré une solubilité très partielle dans les solvants deutérés, nous avons pu réaliser une analyse RMN et confirmer la nature polymérique de ce solide, dont certaines chaines étaient suffisamment courtes pour être solubles.
D'après ces informations, nous avons proposé que le polymère insoluble est formé à partir des alkylations de Friedel-Crafts intermoléculaires successives, dont les carbocations intermédiaires sont générés après abstraction d'un fluorure par donation de liaisons hydrogènes du HFIP. (Schéma 10)
Schéma 10. Mécanisme et structure du produit proposés pour la formation du polymère insoluble lors de la mise en solution du fluorure benzylique dans le HFIP.
Concernant l’utilisation du TFE comme solvant, une plus haute température de réaction a été nécessaire pour que l'abstraction du fluorure se réalise et dans ce cas le produit de solvolyse est formé. (Schéma 11). Nous pouvons penser dans ce cas, que le TFE est un meilleur nucléophile que le HFIP et va donc rapidement réagir avec le carbocation possiblement formé au lieu de le stabiliser.26
Schéma 11. Réaction de solvolyse
En parallèle à la réaction d'amination, nous avons cherché à exploiter la réactivité inattendue des fluorures benzyliques en présence du HFIP. Nous devions notamment trouver un moyen de la contrôler pour empêcher la polymérisation et c'est dans ce contexte que mon projet concernant la réaction de Friedel-Crafts a pris naissance.
2. La réaction de Friedel-Crafts (S
N1)
La réaction de Friedel-Crafts est une réaction de substitution électrophile aromatique, développée en 1877 par le chimiste français Charles Friedel et son partenaire américain James Crafts.27Cette réaction permet de fonctionnaliser des cycles aromatiques par diverses
chaines alkyles ou acyles.
2.1 Alkylation de Friedel-Crafts
Dans la réaction de Friedel-Crafts, le nucléophile est un cycle aromatique, souvent activé par des groupements électro-donneurs. Dans le cas plus précis de l'alkylation, l'électrophile est habituellement un carbocation créé à partir d'un halogénoalcane en présence d'acides de Lewis ou de Brønsted forts.
Le mécanisme réactionnel est présenté ci-dessous. Il commence par l’activation de l’halogénure d’alkyle par un acide de Lewis, provoquant le départ de l’halogène et la formation du carbocation. Celui-ci est piégé par le cycle aromatique qui joue le rôle de nucléophile. L’intermédiaire ainsi formé se réaromatise suite à la perte d’un proton pour former le produit désiré. (Schéma 12)
Schéma 12. Alkylation de Friedel-Crafts
L’échelle présentée dans la Figure 3 classe les substituants du cycle aromatique nucléophile en fonction de leur caractère donneur ou attracteur. Les groupements donneurs comme un éther ou ester favorisent la réaction de Friedel-Crafts et l'attaque de l'électrophile en position ortho/para.28
Les groupements attracteurs comme le trifluorométhyle ou les cétones sont moins favorables à la réaction et l’attaque se fait majoritairement en méta.
27 Friedel, C.; Crafts, J. C. r. hebd. séances Acad. sci. 1877, 84, 1932. 28 Schafer, G.; Bode, J. W. Angew. Chem. Int. Ed. 2011, 50, 13.
Figure 3. Classement des substituants en fonction de leur caractère donneur et attracteur.
La réaction de Friedel-Crafts permet de créer une liaison entre un carbone aliphatique et un carbone aromatique. C'est donc une réaction particulièrement intéressante ce qui est confirmé par le grand nombre de publications sur ce sujet depuis 1978.29 (Figure 4)
Figure 4. Nombre de publications sur la Friedel-Crafts de 1928 à 2007.
29 Bandini, M. General aspects and historical background. In Catalytic asymmetric Friedel-Crafts Alkylations;
Umani-Ronchi, A., Eds.; VCH: Weinheim. 2009.
2.2 Benzylation de Friedel-Crafts :
Dans le cas où le carbocation est formé en position benzylique d'un cycle aromatique du partenaire électrophile, on réalise alors une benzylation de Friedel-Crafts (Schéma 13).
Schéma 13. Schéma général de la benzylation de Friedel-Crafts
Il existe de nombreuses méthodes dans la littérature pour effectuer cette réaction, en voici quelques exemples.
Les médiateurs les plus utilisés pour la formation de carbocations à partir d’halogénures benzyliques sont des acides de Lewis, notamment le AlCl3 et le BF3.
Un des premiers exemples de benzylation de Friedel-Crafts a été développé par Olah en 1972. Ce chercheur est considéré comme le pionnier de la chimie des carbocations.30
(Schéma 14)
Schéma 14. Benzylation de Friedel-Crafts en présence de chlorure d'aluminium (AlCl3)
Dans cette réaction, il utilise un acide de Lewis, le chlorure d'aluminium AlCl3, pour activer
la liaison C-Cl et générer le carbocation suivi par l’attaque du nucléophile, le benzène, formant le produit désiré.
Il existe d'autres précurseurs que les composés halogénés pour la génération des carbocations. Par exemple dans le Schéma 15, le groupement hydroxamate est activé par
le trifluorure de bore en présence du nucléophile, dans ce cas le toluène, pour former le produit désiré à 99% de rendement.28
Schéma 15. Benzylation de Friedel-Crafts en présence de trifluorure de bore (BF3)
Un autre exemple de précurseur de carbocation, le groupement acétate activé par le trichlorure de fer. En présence de o-xylène comme solvant, le produit de substitution est formé avec 99% de rendement (Schéma 16).31
Schéma 16. Benzylation de Friedel-en présence de trichlorure de fer (FeCl3)
D’autres méthodes emploient des métaux de transition comme l’or ou l'iridium pour coordonner le groupement acétate (groupement partant doit être riche en électron) afin de favoriser son départ (Schéma 17).32,33
31 lovel, I.; Mertins, K.; Kischel, J.; Zapf, A.; Beller, M. Angew. Chem. Int. Ed. 2004, 117, 242. 32 Mertins, K.; Iovel, I.; Kischel, J.; Zapf, A.; Beller, M. Adv. Synth. Catal. 2006, 348, 691. 33 Mertins, K.; Iovel, I.; Kischel, J.; Zapf, A.; Beller, M. Angew. Chem. Int. Ed. 2005, 44, 238.
Schéma 17. Benzylation de Friedel-Crafts en présence de métaux de transition
Il est également possible d’utiliser des acides de Brønsted. Dans le cas présenté au Schéma
18, l’acide triflique permet de former un carbocation en position benzylique.34
Schéma 18. Benzylation de Friedel-Crafts en présence d'acide de Brønsted.
2.3 Alkylation de Friedel-Crafts avec des fluorures d'alkyles :
Durant mon projet, je me suis intéressé particulièrement aux fluorures d'alkyle en tant que substrat pro-électrophile. Dans la littérature, il n'existe que peu d'exemples de Friedel-Crafts avec des fluorures d'alkyles comme précurseurs de carbocations.
En 1964, le groupe de Olah a utilisé un acide de Lewis, le AlCl3, pour activer sélectivement
le fluor en présence du benzène.35 Le même type de réaction est réalisé avec du trifluorure
de bore (Schéma 19).36
34 Zhang, Y.; McElrea, A.; Sanchez, G. V.; Do, D.; Gomez, S.; Aguirre, L.; Rendy, R.; Klumpp, D. A. J. Org.
Chem. 2003, 68, 5119.
35 Olah, G. A.; Khun, S. J. Org. Chem. 1964, 29, 2317.
Schéma 19. Activations de fluorures d'alkyle avec des acides de Lewis
Douvris et Ozerov ont utilisé les cations de silicium dans leur réaction afin d'activer un groupement trifluorométhyle. Deux des trois fluors sont remplacés par des hydrogènes et le troisième par un aromatique, le benzène dans cet exemple, pour former le produit à 76% de rendement (Schéma 20).12
Schéma 20. Activation de fluorure d'alkyle en présence de cation silylium catalytique
Un autre exemple d'activation de groupement trifluorométhyle a été effectué en utilisant un acide de Brønsted fort, l’acide triflique. Ce dernier active les trois atomes de fluor du substrat en présence du nucléophile, le benzène, générant majoritairement la cétone, dont l'oxygène provient de l'anion sulfate, à 87% de rendement (Schéma 21).37
Schéma 21. Activation de fluorure d'alkyle par un acide fort de Brønsted
La réactivité des catalyseurs pour l'activation des liaisons C-F peut être évaluée en comparant les forces de liaisons qu'ils créent avec le fluor. Les valeurs sont les suivantes : pour les catalyseurs à base de bore (757 kJ/mol), d'aluminium (664 kJ/mol), de silicium
(553 kJ/mol) et d'hydrogène (570 kJ/mol pour HF, 155 kJ/mol pour les liaisons hydrogènes dans l'ion [HF2]-).38
L'utilisation des fluorures d'alkyles ou d'acyles pour la réaction de Friedel-Crafts pourrait avoir un intérêt en synthèse et permettre la fonctionnalisation orthogonale de substrats ou le fluorure serait un groupement protecteur, devenant partant dans des conditions qui lui sont spécifiques, donnant accès à des molécules de plus en plus complexes.
Comme mentionné précédemment, très peu d'exemples d'utilisation de ces motifs ont été rapportés dans la littérature, car l'intérêt d'activer des fluorures pour réaliser la réaction de Friedel-Crafts est faible face à la grande facilité d'activer plutôt des chlorures ou des bromures. De plus, à l'exception des cations silylium, tous les catalyseurs mentionnés dans les réactions ci-dessus ont d’importants inconvénients. Les acides de Lewis forts à base de bore, d'aluminium ou autre sont très peu sélectifs. Ils ont une affinité plus grande pour n'importe quelle base de Lewis plus forte que l'halogène de l'halogénoalcane. Ils sont alors inactifs lorsqu'ils sont utilisés en présence de phénols ou d'anilines libres.39
L'acide triflique est un acide très fort et très corrosif (donc très difficile d'utilisation) qui met en péril un grand nombre de fonctionnalités possiblement présentes sur l'un ou l'autre des partenaires de la réaction. Également, l'ACS Green Chemistry Institute a identifié la réaction de Friedel-Crafts comme étant l'une des réactions les plus couramment utilisées et pour laquelle de meilleurs réactifs seraient souhaitables.40
Une méthode douce d'activation des fluorures d'alkyles serait intéressante puisqu'elle permettrait éventuellement d'utiliser systématiquement ces motifs lors de la synthèse de molécules complexes.
En conclusion, nous avons vu des exemples de benzylation de Friedel-Crafts sans fluorure et des alkylations de Friedel-Crafts avec des fluorures d’alkyles. Dans mon projet, nous avons décidé de jumeler les deux et d’activer des fluorures benzyliques. Ce type de réaction, n’étant pas décrit dans la littérature, demandera une optimisation minutieuse de plusieurs paramètres pour être contrôler, notamment le choix de substrat et du co-solvant, la
38 Lide, D. R. CRC handbook of chemistry and physics : a ready-reference book of chemical and physical
data; 89éme éd.; CRC Press, London, 2007.
39 Smith, M.; March, J. March's advanced organic chemistry : reactions, mechanisms and structure; 6éme éd.;
Wiley-Interscience : Hoboken, N. J., 2007.
40 Constable, D. J. C.; Dunn, P. J. D.; Humphrey, G. R.; Leazer, J. J. L.; Linderman, R. J.; Lorenz, K.;
concentration et la température. Ces expériences vont nous permettre de tirer de meilleures conclusions et éventuellement un éclaircissement concernant le mécanisme d'activation. 2.4 Objectif du projet
Le but du projet est de contrôler la réaction de Friedel-Crafts initialement observée comme réaction parasite lors des travaux précédents sur l'amination de fluorures de benzyle (voir 1.5). Ce contrôle sera obtenu en utilisant un donneur de liaison hydrogène (DLH) comme activateur de la liaison C-F, après optimisation de la réaction au niveau du solvant, de la température et du nombre d'équivalents. Le but ultime est de développer des conditions favorables pour la substitution nucléophile de fluorures benzyliques dans un milieu neutre. Également, différents substrats et nucléophiles seront testé afin d'élargir l'étendue de la réaction. Enfin, ces optimisations pourront nous aider à comprendre le mécanisme de la réaction. (Schéma 22)
Schéma 22. Optimisation des conditions de la réaction
Les résultats obtenus durant ces travaux, dont une partie a été publiée, seront décrit dans ce mémoire.
3. Synthèse de fluorures de benzyles
3.1. Intérêt des fluorures de benzyles
On sait que les réactions de type SN1 sont très favorisées dans le cas des halogénoalcanes
tertiaires. En ce qui concerne les halogénoalcanes secondaires, les mécanismes SN1 et SN2
sont tout les deux envisageables et dépendent notamment des conditions opératoires. Toutefois, en position benzylique, le carbocation est très stablisé et orientera la réaction vers un mécanisme SN1. Comme mentionné au paragraphe 1.5, il a été démontré que la
liaison C-F benzylique est très réactive en condition d'activation par liaison hydrogene. Ainsi, nous avons choisi le 1-(tert-butyl)-4-(fluorméthyl)benzène comme substrat-modèle. Premièrement, le groupement tert-butyle peut limiter les risques de polymérisation par son encombrement stérique. Deuxièmement, son effet inductif électro-donneur peut favoriser la réaction en stabilisant le carbocation.
3.2. Synthèses des substrats
Les fluorures de benzyles sont une classe de composés très peu décrite dans la littérature. Étant indisponibles sur le marché, nous avons dû les synthétiser selon les réactions suivantes :
3.2.1. Par fluoration de bromures de benzyles :
En fonction de la disponibilité des produits de départ nous exécuterons les réactions suivantes : une réduction de l’aldéhyde avec NaBH4 afin d’obtenir l'alcool benzylique
correspondant, suivie d'une bromation, ou bien effectuer directement la bromation avec la triphénylphosphine en présence de brome si l'alcool est disponible commercialement (Schéma 23).4
Une fois le produit de bromation obtenu, on effectue la fluoration en utilisant le fluorure de tétrabutylammonium trihydrate dans l’acétonitrile pour obtenir le produit final désiré
(Schéma 24).4
Schéma 24. Étape finale de fluoration
3.2.2. Par déoxofluoration :
Dans les cas où les composés bromés sont instables, on effectue la réaction de déoxofluoration qui a pour principe de passer directement de l'alcool au fluor en présence du DAST (Schéma 25).41
Schéma 25. Réaction de déoxofluoration
Après l’obtention des substrats, nous avons décidé d’étudier l’étendue de la réaction. Mon choix initial de nucléophile s’est porté sur le 1,4-diméthoxybenzène, car en plus d'être peu dispendieux, les groupements méthoxy enrichissent le cycle et augmente sa nucléophilie. De plus, il ne pose pas de problème de régioisomérie dans la réaction de Friedel-Crafts.
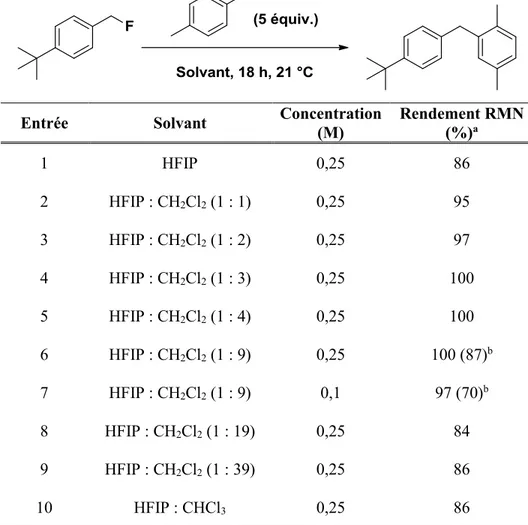
3.3 Optimisation des conditions de la réaction
3.3.1 Optimisation préliminaire: cas du 1,4-diméthoxybenzène
Dans un premier temps, nous avons cherché à déterminer le nombre d'équivalents optimal de nucléophile. Ainsi, tel qu’illustré dans le Tableau 2, la réaction a été faite en utilisant
41 (a) Bio, M. M.; Waters, M.; Javadi, G.; Song, Z. J.; Zhang, F.; Thomas, D. Synthesis 2008, 891. (b)
deux, cinq et dix équivalents du 1,4-diméthoxybenzène, dans le HFIP à température pièce (entrées 1 à 3).
On constate qu’avec deux équivalents on obtient 48% de rendement RMN, ce dernier est obtenu par ajout d’un standard interne ( 2-fluoro-4-nitrotoluène) après le traitement de la réaction. On remarque que si l'on augmente à cinq équivalents, le rendement RMN est augmenté à 86% et baisse légèrement à 83% avec 10 équivalents. Une diminution de la concentration à 0.1 M a conduit à une chute du rendement à 67%, nous avons donc conservé une concentration de 0.25 M.
Étant donné le prix élevé du HFIP (25 g de HFIP coûtent 149,50 $ chez Sigma Aldrich, Août 2015), nous avons décidé de diluer la réaction avec un autre solvant.
Notre choix s'est porté sur le dichlorométhane qui est un solvant non protique et donc n'interfèrera pas dans le mécanisme d'activation par liaison hydrogène. Différents ratios ont été testés (entrées 6 à 13). De façon très intéressante, nous avons remarqué que globalement, la dilution du HFIP par un autre solvant ne s'est pas avérée néfaste. Le rendement est même augmenté à 93% à un ratio de 1 : 1 (entrée 7), puis stagne à 87% jusqu'à un ratio de 1 : 9 (entrée 11). Une diminution trop importante de la quantité de HFIP engagée dans la réaction est par contre rédhibitoire et empêche toute conversion du produit de départ (entrées 12, 13). En remplaçant le HFIP par le TFE (entrées 14 à 17), une nette baisse des rendements est observée et la conversion est même nulle pour le ratio TFE : CH2Cl2 (1 : 2) (entrée 17). Même en chauffant la réaction à 50 °C, seul un modeste 67% de
rendement RMN est atteint (entrée 16). Ce résultat confirme les donnés de l'echelle α indiquant que le TFE est un moins bon donneur de liaison hydrogène que le HFIP.
Le faible rendement qu'on observera tout le long des réactions testées est dû à la formation d'un polymère.
Tableau 2. Optimisation du nombre équivalents avec le 1,4-diméthoxybenzène
Entrée Condition Équivalents Rendement RMN (%)a
1 HFIP, 0.25 M 2 48 2 HFIP, 0.25M 5 86 3 HFIP, 0.25 M 10 83 4 HFIP, 0,1 M 5 67 5 HFIP : CH2Cl2 (1 : 1) 0,25 M 5 93 6 HFIP : CH2Cl2 (1 : 2) 0,25M 5 87 7 HFIP : CH2Cl2 (1 : 3) 0,25 M 5 87 8 HFIP : CH2Cl2 (1 : 4) 0,25 M 5 87 9 HFIP : CH2Cl2 (1 : 9) 0,25 M 5 87 10 HFIP : CH2Cl2 (1 : 19) 0,25 M 5 0b 11 HFIP : CH2Cl2 (1 : 39) 0,25 M 5 0 b 12 TFE, 0,25 M 5 36 13 TFE : CH2Cl2 (1 : 1) 0,25 M 5 43 14 TFE : CH2Cl2 (1 : 1) 0,25 M, 50 °C 5 67 15 TFE : CH2Cl2 (1 : 2) 0,25 M 5 0 b
a: Rendement RMN calculé avec le 2-fluoro-4-nitrotoluène, b : la réaction n’a pas fonctionnée (trés peu de conversion).
Malheureusement, et ce malgré les bons rendements RMN obtenus, nous étions dans l'incapacité de déterminer un rendement isolé. En effet, des problèmes de purification causés par les polarités identiques du 1,4-diméthoxybenzène et du produit de substitution ont rendu l'isolation du produit extrêmement laborieuse. En dépit des efforts fournis pour améliorer les conditions réactionnelles, ce problème de purification n'a pu être surmonté. Nous nous sommes alors tournés vers le p-xylène comme nucléophile et avons répété notre optimisation.
3.3.2 Optimisation en présence de p-xylène Nombre d’équivalents du nucléophile
Nous avons reproduit les conditions du Tableau 2 en remplaçant le 1,4-diméthoxybenzène par le p-xylène qui possède sensiblement les mêmes propriétés. (Tableau 3).
La réaction s’est avèré plus efficace. En effet, une augmentation d'au moins 10% est observée par rapport à l'utilisation du 1,4-diméthoxybenzène. De plus, un rendement quantitatif de 98% est obtenu à cinq équivalents de nucléophile. Nous avons donc conservé le nombre de cinq équivalents pour la suite de notre étude.
Tableau 3. Optimisation du nombre d’équivalents avec le p-xylène
Nombre
d'équivalents Rendement RMN (%)
2 79
5 98
Solvant
Nous sommes ensuite passé à l’optimisation du solvant. Les résultats sont affichés dans le
Tableau 4.
Tableau 4. Optimisation du solvant dans la réaction de substitution nucléophile
Entrée Solvant Concentration (M) Rendement RMN (%)a
1 HFIP 0,25 86 2 HFIP : CH2Cl2 (1 : 1) 0,25 95 3 HFIP : CH2Cl2 (1 : 2) 0,25 97 4 HFIP : CH2Cl2 (1 : 3) 0,25 100 5 HFIP : CH2Cl2 (1 : 4) 0,25 100 6 HFIP : CH2Cl2 (1 : 9) 0,25 100 (87)b 7 HFIP : CH2Cl2 (1 : 9) 0,1 97 (70)b 8 HFIP : CH2Cl2 (1 : 19) 0,25 84 9 HFIP : CH2Cl2 (1 : 39) 0,25 86 10 HFIP : CHCl3 0,25 86
a : Rendement RMN calculé avec le 1,4-diméthoxybenzène comme standard interne, b : Rendement isolé.
Nous avons commencé par réaliser la réaction dans 100% HFIP à une concentration de 0,25 M et nous avons obtenu 86% de rendement RMN. En diluant le HFIP avec le dichlorométhane, nous avons observé une même tendance qu'avec le 2,4-diméthoxybenzène, à savoir une amélioration du rendement. Plus particulierement, en utilisant un ratio HFIP : CH2Cl2 (1 : 9) (entrée 6) nous avons pu isoler le produit de
Friedel-Crafts avec un rendement de 87% équivalent au rendement obtenu avec le HFIP seul. Une diminution de la concentration (entrée 7) ou une augmentation de la dilution du HFIP (entrées 8, 9) n'ont pas permis d'améliorer les conditions, une légère baisse a même été
observée. Malgré les bons rendements RMN obtenus en présence de très faibles quantités de HFIP (entrées 8, 9), nous avons retenues un ratio de HFIP : CH2Cl2 (1 : 9), 0,25 M pour
évaluer l’étendue de la réaction. Finalement, le remplacement du dichlorométhane par le chloroforme n'a pas apporté d'amélioration significative (entrée 10).
Comme dans le cas du 1,4-diméthoxybenzène, nous avons ensuite comparé le HFIP au TFE (Tableau 5). Alors qu'en présence de TFE seul le rendement RMN obtenu est équivalent a celui donné par le HFIP, une dilution du TFE par le CH2Cl2 a conduit à une
chute drastique du rendement qui est passé à 43% (entrée 2) et même à une perte totale de réactivité au ratio (1 : 2) (entrée 3). La moins bonne efficacité du TFE est confirmée, c'est donc le HFIP qui a été retenu pour étudier l'étendue de la réaction.
Le temps de réaction et la température n’ont pas été optimisés pour des raisons pratiques, les rendements obtenus étant très satisfaisant.
Tableau 5. Optimisation de la réaction avec le chloroforme
Entrée Solvant Concentration
(M) Rendement RMN (%)a 1 TFE 0,25 96 2 TFE : CH2Cl2 (1 : 1) 0,25 43 3 TFE : CH2Cl2 (1 : 2) 0,25 0
a: Rendement RMN calculé avec le 2-fluoro-4-nitrotoluène comme standard interne.
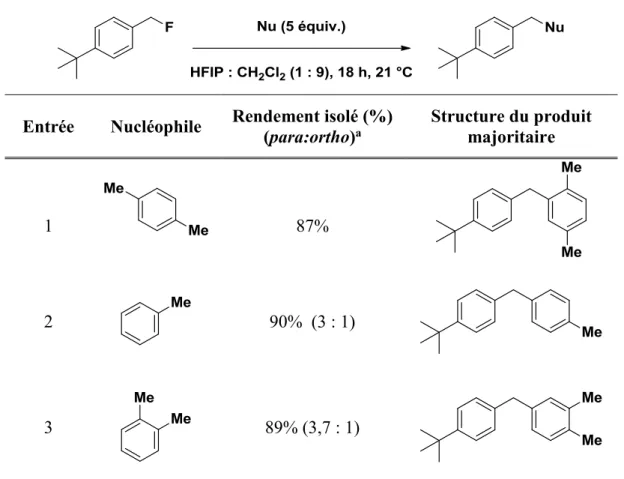
3.4 Étendue de la réaction de substitution nucléophile
Après avoir déterminé les conditions optimales, nous avons cherché à évaluer l’étendue de la réaction. D’abord, nous avons fait varier le nucléophile en testant différents composés aromatiques tels que présentés dans les figures suivantes.
Pour commencer, nous avons testé nos conditions optimales sur des dérivées de diméthoxy et triméthoxybenzène afin de tenter d'obtenir de meilleurs résulats que lors de l'optmisation préliminaire du 1,4-diméthoxybenzène. Ainsi, nous avons testé nos conditions optimales (CH2Cl2 : HFIP (9 :1), 0,25 M à 21 °C sur 18 h) avec le 1,3-diméthoxybenzène, le
Figure 5. Réaction test avec différents nucléophiles substitué avec des groupements méthoxy
Malheureusement, les mêmes problèmes de purification rencontrés dans le cas du 1,4-diméthoxybenzène ont été constatés et cela malgré un rendement RMN supérieur à 70%. En revanche, l’anisole dans les mêmes conditions a permis d'obtenir un rendement isolé de 87%, la purification n'étant pas problématique dans ce cas. (Schéma 28)
Schéma 26. Réaction avec l’anisole
On remarque clairement la présence des deux régioisomère avec un ratio para : ortho de (1,4 : 1). La régioisomérie a été déterminé par RMN proton en comparant avec les données de la littérature.42
Nous avons également testé la réaction avec les aromatiques contenants des substituants méthyles (Tableau 6). Contrairement aux groupements méthoxy, aucun problème de purification n'a été rencontré et les résultats étaient très satisfaisants. L’effet inductif donneur du groupement méthyle sur l’aromatique favorise l’attaque sur les positions ortho et para. Ainsi, nous avons obtenu un rendement de 87% avec le p-xylène (entrée 1), 90% avec le toluène (entrée 2) avec un ratio (3 : 1) et un 89% avec l’o-xylène avec un ratio de (3,7 : 1), toujours en faveur du produit para substitué.
Tableau 6. Étendue de la réaction de Friedel-Crafts avec des groupements méthyles
Entrée Nucléophile Rendement isolé (%) (para:ortho)a Structure du produit majoritaire
1 87%
2 90% (3 : 1)
3 89% (3,7 : 1)
a : le ratio p : o a été déterminé par RMN 1H.
En ce qui concerne, les aromatiques non substitués (Tableau 7), il a été nécessaire d’effectuer des ajustements de nos conditions optimales. En effet, dans le cas du naphtalène (entrée 1) on obtient 26% de rendement isolé avec HFIP : CH2Cl2 (1 : 9). En modifiant le
ratio de HFIP : CH2Cl2 à (9 : 1) et en présence de 10 équivalents de naphtalène, nous avons
isolé le produit à 80% de rendement (entrée 2). Le produit majoritairement obtenu est substitué en position 1 avec un ratio de (2,8 : 1).
Dans le cas du benzène, on observe également une conversion complète, mais on ne récupère que 26% (entrée 3) de rendement avec HFIP : CH2Cl2 (1 : 9). Là aussi, nous avons
augmenté le ratio HFIP : CH2Cl2 et le nombre d’équivalents de benzène permettant cette
Suite aux résultats peu concluants obtenus dans le cas du benzène, nous avons pensé à une autre approche qui consiste à remplacer le solvant (CH2Cl2) par le nucléophile lui-même
(benzène). Ainsi, le nombre d’équivalents de nucléophile est augmenté de façon conséquente et cela nous a permi d’améliorer le rendement, comme nous le montre (l’entrée 5) où l’on obtient un rendement de 57% . Ces résultats plus faibles sont cohérents avec la faible nucléophilie de ces réactifs.
Tableau 7. Étendue de la réaction de Friedel-Crafts avec des nucléophiles non substitués.
Entrée Nucléophile Conditions Ratioa Rendement isolé (%) Structure 1 HFIP : CH2Cl2 (1 : 9), 5 équiv. - 26% 2 HFIP : CH2Cl2 (9 : 1) 10 équiv. (2,8 : 1) 80% 3 HFIP : CH2Cl2 (1 : 9), 5 équiv. - 26% 4 HFIP : CH2Cl2 (9 : 1) 10 équiv. - 43% 5 HFIP : PhH (1: 9) 57%
a: le ratio position 1 : position 2 a été déterminé par RMN 1H.
La réaction a également été testée avec des hétérocycles aromatiques (Tableau 8). Pour ce genre de réactif, nous avons effectué plusieurs tests dans différentes conditions en fonction de la nucléophilie de chaque hétérocycle afin d’obtenir les meilleurs rendements.
Dans le cas du 2-méthylfurane, des produits secondaires de polymérisations nous ont empèchés d’obtenir un rendement isolé, d’où les rendements RMN mentionnés (entrée 1 et 2). Ces résultats peuvent s’expliquer par la faible nucléophilie du 2-méthylfurane ne favorisant pas la réaction souhaitée, mais plutôt l'attaque du réactif sur lui-même.
Pour le thiophène, nous n'avons obtenu que 40% de rendement isolé dans nos conditions initiales, avec un ratio de (2,4 : 1) en faveur de l'attaque en position 2 (entrée 3). Là encore, afin d'obtenir un meilleur rendement nous avons modifié les conditions en augmentant le
ratio HFIP : CH2Cl2 à (9 : 1) avec 10 équivalents de nucléophile. De cette manière le
produit a été isolé à 90% avec un ratio de (3 : 1) en position 2 (entrée 4).
Par contre, dans le cas du N-méthylpyrrole, les faibles rendements de 47% et de 54% obtenus dans les conditions initiales (entrée 5 et 6), nous ont conduit a utiliser des conditions plus drastiques. Ainsi le DCM a été remplacé par le DCE nous permettant de chauffer la réaction à 60 °C et l’obtention d’un rendement de 80% (entrée 7). On remarque dans ce cas, que la sélèctivité des attaques est plus faible mais toujours en faveur de la position 2 du N-méthylpyrrole.
Tableau 8. Étendue de la réaction de Friedel-Crafts avec des hétérocycles aromatiques
Entrée Nucléophile Conditions Ratioa Rendement RMN (%) Rendement isolé (%) Structure 1 HFIP : CH2Cl2 (1 : 9), 5 équiv. - 26% - 2 HFIP : CH2Cl2 (9 : 1), 5 équiv. - 43% - 3 HFIP : CH2Cl2 (1 : 9), 5 équiv. 2,4:1 56% 40% 4 HFIP : CH2Cl2 (9 : 1), 10 équiv. 3 : 1 - 90% 5 HFIP : CH2Cl2 (1 : 1), 5 équiv. 1,1:1 - 47% 6 HFIP : CH2Cl2 (1 : 1) 10 équiv. 1,2:1 - 54% 7 HFIP : C2H4Cl2 (1 : 9) 5 équiv., 60 °C 1,3 : 1 - 80%
Ensuite, nous avons testé des benzènes substitués par des groupements halogénés. L'utilisation de nucléophiles trifluorométhylé et chlorés n'a conduit qu'à des réactions secondaires de polymérisations ne permettant pas d'isoler les produits désirés (entrée 1 à 4)
(Tableau 9). Seul le fluorobenzène a donné un rendement RMN moyen de 53% (entrée 5).
Ces résultats s'expliquent par la présence des groupements inductifs attracteurs qui ne favorisent pas l’attaque du nucléophile puisqu'ils appauvrissent le cycle aromatique en électrons. Il y a donc compétition avec la nucléophilie du cycle benzénique du substrat qui peut réagir sur lui même et là encore conduire a une réaction de polymérisation. Toutefois, dans le cas du fluorobenzène, l'effet mésomère donneur du fluor annule son effet inductif attracteur ce qui lui confère une réactivité proche de celle du benzène expliquant le rendement de 53%.
Tableau 9. Étendue de la réaction de Friedel-Crafts avec des substituants halogénés
Entrée Nucléophile Conditions Rendement RMN (%) Rendement isolé (%) 1 HFIP : CH2Cl2 (1 : 9), 5 équiv. - 5% 2 Polymère - 3 HFIP : CH2Cl2 (1 : 9), 5 équiv. Polymère - 4 HFIP : CH2Cl2 (9 : 1), 10 équiv. Polymère - 5 HFIP : CH2Cl2 (1 : 9), 5 équiv. 53% -
Il faut savoir que dans tous les tests effectués avec les nucléophiles décrits précédemment la conversion était complète. Cependant, la formation d'impuretés probablement polymériques a conduit dans certains cas à une baisse de rendement par consommation des réactifs dans ces réactions secondaires. Dans l'ensemble, ces résultats montrent que tant que la nucléophilie de l'arène est supérieure à celle du fluorure benzylique ou quand le nucléophile est présent en excès, la réaction de Friedel-Crafts souhaitée peut être observée à la place de la polymérisation.
Enfin, nous avons engagé des nucléophiles benzéniques substitués par des groupements carbonylés élèctrodonneurs tels que un acétate (entrée 1,2,3), un acétamide (entrée 4), ou élèctroattracteurs tels que un ester éthylique (entrée 5 à 8) et un aldéhyde (entrée 9 à 10).
Malgré la variété de conditions testées et mêmes un chauffage à 60 °C dans le cas de l'aldéhyde (entrée 11) les produits désirés n'ont jamais été observés par RMN (Tableau 10). Le mécanisme de réaction peut nous permettre de comprendre ces résultats. En effet, les substituants carbonylés dont l’acétate sont de meilleur accepteur de liaison hydrogène, que le substrat fluoré et donc le HFIP va favoriser sa coordination avec le nucléophile au détriment du substrat justifiant le 0% de conversion.
Tableau 10. Étendue de la réaction de Friedel-Crafts avec des substituants carbonylés
Entrée Nucléophile Conditions
1 HFIP : CH2Cl2 (1 : 9), 5 équiv 2 HFIP : CH2Cl2 (1 : 1), 5 équiv 3 HFIP : CH2Cl2 (9 : 1), 5 équiv 4 HFIP : CH2Cl2 (1 : 9), 5 équiv 5 HFIP : CH2Cl2 (1 : 9), 5 équiv 6 HFIP : CH2Cl2 (1 : 1), 5 équiv 7 HFIP : CH2Cl2 (1 : 9), 2 équiv 8 HFIP : CH2Cl2 (1 : 1), 0,5 équiv 9 HFIP : CH2Cl2 (1 : 1), 5 équiv 10 100% HFIP 11 HFIP : C2H4Cl2 (1 : 9) 5 équiv, 60 °C
J’ai également essayé d’autres nucléophiles tels que l’indole, le ferrocène, le naphtol et le méthoxynaphtalène, mais aucun produit désiré n'a été détécté par RMN. Les réactions n'ont pas été poussées plus que ça.