Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Chimie organique, minérale, industrielle
Présentée par :
Alexandre Le Darz
Activation superélectrophile en milieu superacide :
synthèse de nouveaux synthons halofluorés azotés
Directeur(s) de Thèse :
Sébastien Thibaudeau, Agnès Martin-Mingot
Soutenue le 24 novembre 2014 devant le jury
Jury :
Président Danièle Bonnet-Delpon Directeur de recherche CNRS émérite, Université de Paris Sud Rapporteur Thierry Brigaud Professeur des Universités, Université de Cergy-Pontoise Rapporteur Thierry Billard Directeur de recherche CNRS, Université de Lyon Membre Sébastien Thibaudeau Maître de conférences, Université de Poitiers Membre Agnès Martin-Mingot Maître de conférences, Université de Poitiers
Membre Jacques Fahy Conseiller scientifique, Centre de recherche Pierre Fabre, Castres
Membre Yves Blériot Professeur des Universités, Université de Poitiers
Membre Fodil Bouazza Docteur, Société @rtMolécule, Poitiers
Pour citer cette thèse :
Alexandre Le Darz. Activation superélectrophile en milieu superacide : synthèse de nouveaux synthons halofluorés azotés [En ligne]. Thèse Chimie organique, minérale, industrielle. Poitiers : Université de Poitiers, 2014. Disponible sur Internet <http://theses.univ-poitiers.fr>
Pou l’o te tio du g ade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
Faculté des Sciences Fondamentales et Appliquées
(Diplôme national – arrêté du 7 août 2006)
École Doctorale : S ie es pou l’E i o e e t Ga Lussa Secteur de recherche : Chimie Organique, Minérale et Industrielle
Présentée par :
Alexandre LE DARZ
¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤
ACTIVATION SUPERELECTROPHILE EN MILIEU SUPERACIDE :
SYNTHESE DE NOUVEAUX SYNTHONS HALOFLUORES AZOTES.
¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤
Directeurs de thèse :
Sébastien Thibaudeau, Maître de conférences (HDR), Université de Poitiers Agnès Martin-Mingot, Maître de conférences, Université de Poitiers
¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤ Soutenue le 24 novembre 2014 De a t la o issio d’e a e ¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤¤ JURY Pr Thierry BRIGAUD, Dr Thierry BILLARD, Pr Danièle BONNET-DELPON, Dr Jacques FAHY, Dr Fodil BOUAZZA, Pr Yves BLÉRIOT Dr Sébastien THIBAUDEAU, Dr Agnès MARTIN-MINGOT,
P ofesseu à l’université de Cergy Pontoise Di e teu de e he he CNRS à l’université de Lyon
Directeur de recherche CNRS émérite à l’u i e sit de Pa is Sud
Co seille s ie tifi ue da s l’u it de se i es de recherche au centre de recherche et déve-loppement Pierre Fabre, Castres
Responsable recherche et développement SARL @rtMolecule, Poitiers
P ofesseu à l’u i e sit de Poitie s
Maît e de o f e es à l’u i e sit de Poitie s Maît e de o f e es à l’u i e sit de Poitie s
Rapporteur Rapporteur Examinateur Examinateur Examinateur Examinateur Examinateur Examinateur
ner à bien ce projet, autant par leur présence au quotidien à mes côtés au laboratoire que
par leur soutien moral pendant ces trois années.
Je remercie les Dr Sébastien Thibaudeau et Agnès Mingot pour leur disponibilité, leur
écoute, leur sympathie et tous les o s o seils ui ’o t guid tout au lo g de ette th se.
Sa s eux e t avail ’au ait pas pu a outi .
Je remercie également les docteurs Fabien Zunino, Omar Karam, et Fodil Bouazza de
’avoi do
l’oppo tu it de alise e t avail. L’a ueil toujou s haleu eux et l’o eille
toujours attentive, ils ont su rester présents et toujours de bons conseils pour faire avancer
es t avaux. Me tio sp iale à Fodil ui ’a ja ais h site à p e d e su so te ps pou
venir discuter avec moi
ua d j’e avais besoin, que ça soit des travaux de thèse ou de la vie
en général. Merci pour ce précieux soutien.
Un grand merci à Ornicar, Poussin, Hélène et Longin pour la bonne humeur qui a
ré-gné dans le laboratoire. Les manips qui foirent se digèrent mieux dans un
li at d’e te te et
de complicité. Merci à eux de ’avoi suppo t pe da t t ois ans. Mention spéciale à poussin
et Ornicar pour les week-ends passés e se
le et l’aide u’ils ’o t vailla
e t appo t e
pour la sauvegarde du palais de la bière.
Au tour du club des fines gueules, Mikael, Taz, Fatima, Hannitra, Ugo, Kavita, Lapin,
Nadège pour le sanglier, le cerf, les truites, le St Émilion et aut es de
es ui ’o t pe is
de su siste . Bie a ge ’est le d ut du o heu , e o e o pag ie ça ’a pas de prix.
Je n’ou lie pas o plus la du-genou team, Romain, suricate, taz-bis, la touffe et
Mi-kael, pour leur accueil à mon arrivée sur Poitiers et toutes les soirées passées ensemble
de-puis. Mention spéciale à Soizic pour mon
a e ue… e tio sp iale au suricate pour les
ideaux… et e tio sp iale au eilleu MJ du o de.
Merci aussi à tous les membres du plb, du club de fléchettes, et aux amis du poker
pour tous les bons moments passés en leur compagnie
U g a d e i à es a is d’e fa e Pik et Te ui ont fait plusieurs fois le long
tra-jet qui nous sépare.
E fi je tie s à e e ie toute a fa ille pou le soutie i d fe ti le u’elle ’a
ap-po t e du a t es t ois a
es de th se et toutes elles d’ava t.
Abréviations
Ac : acétyl
AcOEt : acétate d’éthyle AcN : acétonitrile
All : allyle Bn : benzyle
Boc : tert-butyloxycarbonyle Bu : butyl
CCM : chromatographie sur couche mince CDP : cytidine-diphosphate
COSY : Correlation Spectroscopy D : debye
d : doublet
DAST : trifluorure de diéthylaminosulfure
DEPT : Distortionless Enhancement by Polarization Transfer DMAP : 4,4-diméthylaminopyridine DMSO : diméthylsulfoxyde DMF : diméthylformamide DRX : diffractométrie de rayon X EP : éther de pétrole éq : équivalent Et : éthyle h : heure
HMBC : Heteronuclear Multiple Bond Correlation HSQC : Heteronuclear Single Quantum Coherence IC50 : concentration d’inhibition à 50%
LDA : diisopropylamidure de Lithium Me : méthyle MeOH : méthanol min : minute Ms : mésyl NBS : N-bromosuccinimide NCS : N-chlorosuccinimide NDP : nucléoside-diphosphate NFSI : N-fluorobenzènesufonimide Ns : Nosyl
NXS : N-halosuccinimide Pf : point de fusion Ph : phényle Phtal : phtalimide ppm : partie par million p-Tol : para toluène Pyr. : pyridine q : quadruplet quant. : quantitatif rel. : relatif
Rf : Rapport frontal
RMN : Résonnance Magnétique Nucléaire s : singulet
SM : spectrométrie de masse
SMHR : spectrométrie de masse haute résolution t : triplet
TA : température ambiante
TBAF : fluorure de tétrabutylamonium
TEP : Tomographie par Émission de Positrons TFA : acide trifluoroacétique
THF : tétrahydrofurane TMS : triméthylsilyle Ts : tosyle
Table des matières
PREAMBULE ... 15
ÉTUDE BIBLIOGRAPHIQUE ... 19
1.
Le fluor et les molécules fluorées ... 21
1.1
Propriétés physico-chimiques ... 22
1.1.1
Électronégativité ... 22
1.1.2
Taille ... 22
1.1.3
Longueur de liaison... 22
1.1.4
Polarisabilité ... 23
1.1.5
Énergie de liaison ... 23
1.2
Effet du fluor dans les composés organofluorés ... 23
1.2.1
Effet gauche ... 23
1.2.2
Interactions dipolaires ... 24
1.2.3
Liaisons hydrogènes... 25
1.2.4
I flue e de la p se e d’ato es de fluo su la sta ilit des i te
diai es
réactionnels. ... 26
1.2.4.1
Radicaux ... 26
1.2.4.2
Carbocations ... 27
1.2.4.3
Carbanions ... 28
1.2.5
I flue e de la p se e d’u ato e de fluo su le pKa ... 28
1.2.6
Lipophilie ... 29
2.
Fluor et chimie médicinale ... 30
2.1
Intérêt des motifs fluorés en chimie thérapeutique. ... 30
2.1.1
Biodisponibilité ... 30
2.1.2
Résistance métabolique ... 31
2.1.3
Biomimétisme ... 32
2.2
Méthodes de synthèse de -fluoroamines ... 33
2.2.1
Fluoration par voie nucléophile ... 34
2.2.1.1
Su stitutio u l ophile d’a i o-alcool ... 34
2.2.1.2
Fluo atio d’al ool e z li ue ... 35
2.2.1.3
Ou e tu e d’azi idi e... 35
2.2.2
Fluoration par voie électrophile ... 37
2.2.2.1
α-fluo atio d’i i es et du tio ... 37
2.2.3
Aminofluoration ... 37
2.2.3.1
Fluoration catalysée par le fer ... 37
2.2.3.2
Aminofluoration de styrène palladocatalysée ... 38
2.2.4
Utilisation de synthons fluorés ... 38
2.2.4.1
Fluoroalcools ... 38
2.2.4.2
Fluoroalkyles ... 38
2.2.4.3
Fluorohalogénés ... 39
2.2.5
Fluoration en milieu superacide ... 40
3.
Les milieux superacides ... 40
3.1
Propriétés des milieux superacides ... 41
3.1.1
Mesu e de l’a idit ... 41
3.1.2
Différents types de superacides ... 41
3.1.3
HF anhydre ... 42
3.1.4
Composition du mélange HF/SbF
5... 43
4.
Les superélectrophiles ... 44
4.1
Superélectrophiles gitoniques et distoniques ... 45
4.2
Superélectrophiles azotés ... 46
4.2.1
Réaction de type Friedel Crafts ... 46
4.2.1.1
Voie intramoléculaire ... 46
4.2.1.2
Voie intermoléculaire ... 47
4.2.2
Hydrofluoration ... 48
4.2.3
Cyclisation/Fluoration... 49
4.2.4
Dications Ammonium/Halonium ... 49
5.
les ions haloniums ... 50
5.1
Ions iodoniums ... 52
5.1.1
Études par spectroscopie RMN ... 52
5.1.2
Conclusion ... 52
5.2
Ions bromonium ... 53
5.2.1
Études par spectroscopie RMN ... 53
5.2.3
Étude par difractométrie de rayon X ... 54
5.2.4
Études théoriques ... 54
5.2.5
Étude par spectrométrie de masse ... 56
5.2.6
Conclusion ... 56
5.3
Ions chloroniums ... 56
5.3.1
Études par spectroscopie RMN ... 57
5.3.2
Étude par spectroscopie infrarouge ... 58
5.3.3
Études théoriques ... 58
5.3.4
Étude par spectrométrie de masse ... 60
5.3.5
Conclusion ... 60
5.4
Ions fluoroniums ... 60
5.4.1
Études par spectroscopie RMN ... 60
5.4.2
Études théoriques ... 62
5.4.3
Étude par spectrométrie de masse ... 63
5.4.4
Étude empirique ... 63
5.5
Conclusion ... 64
RESULTATS ET DISCUSSION ... 67
6.
Réaction de chlorofluoration ... 69
6.1
Contexte ... 69
6.2
Études préliminaires ... 71
Détermination structurale ... 72
Discussion ... 74
6.3
Influence des paramètres expérimentaux ... 74
6.3.1
Acidité du milieu ... 74
6.3.2
Temps de réaction ... 75
6.3.3
Évaluation par RMN in situ à basse température de la protonation de la
N-chlorosuccinimide ... 76
6.3.4
Tests d’ uili es ... 79
6.3.5
Température ... 79
6.3.6
Dilution et quantité de NCS ... 80
6.3.7
É aluatio de la fo
atio possi le d’u i te
diai e a tio el io
chloronium cyclique ... 81
Détermination structurale ... 83
Discussion ... 84
6.3.8
Mécanisme réactionnel ... 84
6.4
Influence de la structure du substrat ... 86
6.4.1
Amines aromatiques ... 86
6.4.1.1
Synthèse des substrats ... 86
6.4.1.2
Étude de la réactivité des substrats ... 86
Discussion ... 88
6.4.2
Amines hétéroaromatiques ... 89
6.4.2.1
Synthèse des substrats ... 89
6.4.2.2
Étude de la réactivité des substrats ... 89
Discussion ... 90
6.4.3
Autres fonctions azotées ... 92
6.4.3.1
Synthèse des substrats ... 92
6.4.3.2
Étude de la réactivité des substrats ... 92
6.4.3.3
Détermination structurale des produits non chlorofluorés ... 93
Discussion ... 95
6.4.4
Réactivité de sulfonamide tertiaires chirales ... 99
6.4.4.1
Détermination structurale ... 101
Discussion ... 103
6.4.5
Influence de la présence de fonctions en système aliphatique et de la
distance intercationique. ... 105
6.4.5.1
Synthèse des substrats ... 105
6.4.5.2
Étude de la réactivité des substrats ... 106
6.4.5.3
Discussion ... 107
6.5
Formation de composés polyhalofluorés ... 108
6.5.1.1
Étude préliminaire ... 108
6.6
Réaction de composés insaturés halogénés ... 112
6.7
Application à des composés de structure complexe ... 114
CONCLUSION GENERALE ... 115
PARTIE EXPERIMENTALE ... 119
7.1
Suivi et purification ... 121
7.2
Analyses et caractérisation des produits obtenus ... 121
7.3
Protocole pour les réactions en milieu superacide ... 122
8.
Réaction de chlorofluoration ... 122
8.1
Synthèse des substrats... 122
8.2
Synthèse des composés azotés fluorés
chlorés ... 124
Procédure générale A ... 124
Procédure générale B ... 124
8.3
Synthèse des composés polyhalogénés ... 158
8.4
Application à des composés de structure complexe ... 165
Les propriétés exceptionnelles des composés fluorés ont entrainé un développement considérable de la chimie du fluor depuis les années 1950. Aujourd’hui les substances fluo-rées se retrouvent dans la plupart des domaines tels que la chimie thérapeutique, agrochi-mie ou la chiagrochi-mie des matériaux.
Le fluor est l’halogène le plus abondant de la croute terrestre, pourtant celui-ci est quasi inexistant dans les molécules organiques naturelles. La chimie médicinale s’inspire largement de la nature dans la découverte de nouvelles substances actives. Ainsi l’introduction d’atomes de fluor dans les produits pharmaceutiques est assez récente. L’intérêt du fluor en chimie médicinale et en agrochimie est désormais largement reconnu, les substances fluorées représentent près d’un quart des parts de marché dans le domaine pharmaceutique et près de la moitié dans le domaine agrochimique.
L’atome d’azote est ubiquitaire en chimie médicinale, l’introduction d’un atome de fluor à proximité de celui-ci induit des modifications des propriétés physicochimiques très intéressantes que ce soit en termes de biodisponibilité ou de résistance métabolique. Le dé-veloppement de méthodes permettant l’obtention de ces composés est donc essentiel. Mal-gré le nombre et la diversité des méthodes de fluoration, l’obtention de composés azotés fluorés reste un challenge en chimie organique.
Une des approches possibles consiste en la préparation de synthons azotés halofluo-rés, présentant un fort potentiel synthétique. Malheureusement la synthèse de ces synthons en milieux dits « conventionnels » est extrêmement difficile.
L’utilisation de milieux superacide modifie fortement la réactivité des substrats orga-niques. Dans ces conditions extrêmes, il est possible d’accroitre la réactivité de certains réactifs ou de désactiver certains groupements fonctionnels par protonation.
Le travail présenté ici vise à développer une méthode de chlorofluoration directe de composés azotés insaturés. L’utilisation des conditions superacides entraîne une activation du donneur d’ion halénium par polyprotonation permettant l’accès à un intermédiaire superé-lectrophile, capable de réagir avec des composés insaturés non activés voire même désacti-vés.
Dans la première partie, quelques généralités sur les caractéristiques du fluor et ses effets sur les composés organiques seront présentés. Les propriétés des milieux supera-cides ainsi que des espèces superélectrophiles générées dans ces conditions seront abor-dées. Enfin la structure et la stabilité des différents ions haloniums seront discutées.
Les résultats obtenus lors du développement de la réaction de chlorofluoration en mi-lieu superacide seront présentés dans la deuxième partie.
Le premier chapitre, est dédié à la mise au point de conditions opératoires permettant la chlorofluoration régiosélective de composés azotés insaturés. L’influence de divers para-mètres expérimentaux sera évaluée. Une étude mécanistique par RMN in situ à basse tem-pérature et par analyse des divers produits obtenus sera également présentée.
L’impact de la structure du substrat sur le cours de la réaction sera étudié dans le se-cond chapitre.
Le troisième chapitre sera consacré à l’étude de la réactivité de substrats haloally-liques dans les conditions de chlorofluoration.
Enfin la méthode développée sera appliquée pour la chlorofluoration directe de com-posés élaborés.
Pour des raisons de confidentialité, une partie des travaux réalisés au cours de ces 3 années de thèse ne sera pas présenté dans ce manuscrit.
1.
Le fluor et les molécules fluorées
La première apparition du terme fluor remonte au 16ème siècle. Sous forme cristalline
de CaF2 appelée « fluorite », il est alors utilisé comme additif permettant de faciliter la fusion
des minerais.
Au 18ème siècle la fluorite est combinée à de l’acide sulfurique formant ainsi l’acide
fluorhydrique utilisé pour le polissage du verre.
Au 19ème siècle de nombreux chimistes ont tenté sans succès d’isoler le fluor F 2
jus-qu’en 1886 où Henri Moissan l’obtient par électrolyse d’acide fluorhydrique et de fluorure de potassium. En 1906 il reçoit le prix Nobel de chimie pour cette découverte.
La chimie du fluor reste anecdotique jusqu’au milieu du 20ème siècle. Par la suite,
l’utilisation de fluor F2 et d’acide fluorhydrique HF par l’industrie thermonucléaire pour
l’enrichissement isotopique de l’uranium conduit à leur production à grande échelle. La chi-mie des matériaux commence à développer des polymères fluorés tel que le Teflon®
com-mercialisé en 1960. Les premières substances bioactives fluorées apparaissent en 1954 avec les dérivés de corticoïdes tels que la bétaméthasone ou la dexaméthasone1 encore
utilisés aujourd’hui comme anti-inflammatoires (Figure 1).
O HO F O OH OH O HO F O OH OH Bétaméthasone Dexaméthasone
Figure 1 : Exemples de corticoïdes fluorés.
Par la suite, le nombre de substances actives contenant un ou plusieurs atomes de fluor a constamment augmenté. Aujourd’hui, 25% des molécules pharmaceutiques contien-nent au minimum un atome de fluor.2
1 J. Fried, E.F. Sabo, J. Am. Chem. Soc. 1954, 76, 1455.
1.1 Propriétés physico-chimiques
1.1.1 Électronégativité
Le fluor est l’élément le plus électronégatif du tableau périodique (Tableau 1). D’une valeur de 3,98 sur l’échelle de Pauling, cette forte électronégativité du fluor induit une forte polarisation de la liaison C-F lui conférant un caractère ionique notable.
Tableau 1 : Électronégativité (χp) des éléments selon Pauling par ordre décroissant.
F O Cl N Br I C H χp 3,98 3,44 3,16 3,04 2,96 2,66 2,55 2,20
1.1.2 Taille
Le rayon de Van Der Waals du fluor est parmi les plus faibles (Tableau 2), il est assez proche des rayons de l’hydrogène et de l’oxygène.
Tableau 2 : Rayon de van der waals (rv) des éléments par ordre croissant.
H Zn He Cu F O Be Ne
rv Ǻ 1,20 1,39 1,40 1,40 1,47 1,52 1,53 1,54
1.1.3 Longueur de liaison
La longueur moyenne de la liaison C-F est proche de celle des liaisons C-H et C-O (Tableau 3).3
Tableau 3 : Longueur de liaison (L C-X) par ordre croissant.
H F O N C Cl S P
L C-X Ǻ 1,10 1,41 1,43 1,47 1,54 1,76 1,81 1,87
1.1.4 Polarisabilité
Cette forte électronégativité et ce faible rayon atomique font du fluor l’atome le moins polarisable hormis les gaz rares (Tableau 4).4
Tableau 4 : Polarisabilité (αd) des éléments par ordre croissant.
F H O N C Cl S Br I αd (Ǻ3) 0,56 0,67 0,80 1,10 1,76 2,18 2,90 3,05 4,70
1.1.5 Énergie de liaison
Le fort caractère ionique de la liaison carbone-fluor associé à la faible polarisabilité du fluor induisent une énergie de liaison carbone-fluor plus forte que celle du carbone avec tout autre atome (Tableau 5).
Tableau 5 : Potentiel d’ionisation (Pi) par ordre décroissant.
F N O H Cl Br C Pi (kcal.mol-1) 401,8 335,1 314,0 313,6 299,0 272,4 240,5
Les caractéristiques exceptionnelles du fluor entraînent de nombreuses modifications des propriétés moléculaires des composés organiques fluorés par rapport à leurs homo-logues non fluorés.
1.2 Effet du fluor dans les composés organofluorés
1.2.1 Effet gauche
La liaison C-F étant fortement polarisée, l’orbitale anti-liante σ* est de faible énergie,
elle peut donc facilement être stabilisée par l’orbitale liante σ d’une liaison C-H voisine. Alors que la plupart des composés adoptent une conformation anti afin de minimiser les interac-tions stériques, cette stabilisation par hyperconjugaison induit une conformation gauche privi-légiée des composés fluorés (Figure 2).5
4 J. K. Nagle, J. Am. Chem. Soc., 1990, 112, 4741. 5 D. O’Haga , Chem. Soc. Rev., 2007, 37, 308.
H H F F H H F H H F H H H F conformation gauche conformation anti
0,0 kcal.mol-1 0,5-0,9 kcal.mol-1
CH CF
Figure 2 : Effet gauche du fluor.
1.2.2 Interactions dipolaires
Le caractère ionique très marqué de la liaison Cδ+-Fδ- entraîne un fort moment
dipo-laire (exemple du fluorométhane µ = 1,85 D). Les interactions dipodipo-laires, qu’elles soient at-tractives ou répulsives, jouent donc un rôle prépondérant dans la conformation des compo-sés fluorés.
Da s l’e e ple i-après, le composé amide fluoré 1a et son homologue méthylé 1b ont été analysés par diffraction des ont été analysés par diffraction des rayons X (
Figure 3).6 La conformation quasi planaire du composé fluoré 1a présentant l’atome
de fluor en position cis par rapport au NH, et en trans du carbonyle implique de fortes inte-ractions dipolaires entre ces différentes fonctions. Le composé non fluoré 1b adopte quant à lui la conformation minimisant les interactions stériques.
RHN O H Me H O H2FNR H H
Figure 3 : Conformations privilégiées déduites des données obtenues par DRX de N-acétyl-(S)-phénylalanine fluorée et non fluorée. 6
Le dipôle formé par la liaison Cδ+-Fδ- interagit encore plus fortement avec les
hétéroa-tomes portant une charge formelle. Cette propriété est particulièrement bien illustrée par les travaux de Lankin et Snyder sur la conformation de pipéridines fluorées protonées (Figure
6 J. W. Banks, A. S. Batsanov, J. A. K. Ho a d, D. O’Haga , H. S. Rzepa, S. Ma ti -Santamaria, J. Chem. Soc.,
Per-kin Trans. 2, 1999, 2409. H N O OH H O R 1a R = F 1b R = Me 1a R = F 1b R = Me
4).7 Dans cet exemple la conformation présentant l’atome de fluor en position axiale est
privi-légiée malgré la gêne stérique qui pourrait en résulter.
N H F H N H F H + + 0,0 kcal.mol-1 5,4 kcal.mol-1
Figure 4 : Énergies relatives de conformères de pipéridines fluorées.
1.2.3 Liaisons hydrogènes
La liaison hydrogène est caractérisée par une distance interatomique C-Y···H-X de l’ordre de 2,3 Ǻ. Malgré trois doublets non liants et une forte électronégativité, le fluor a long-temps été considéré comme un mauvais accepteur de liaisons hydrogènes. Depuis quelques années de nombreux auteurs ont cherché à démontrer l’existence de liaisons hydrogènes impliquant un atome de fluor comme accepteur. Hans-Jörg Schneider8 a récemment reporté
les travaux les plus récents donnant ainsi un aperçu de l’état de l’art en la matière.
Finalement il semblerait que le fluor puisse jouer le rôle d’accepteur avec de forts donneurs de liaison hydrogène tel que H2O ou HF. En revanche lorsque le donneur est plus
faible il devient difficile de distinguer si le rapprochement du fluor et de l’hydrogène est dû à une liaison hydrogène, à d’autres interactions faibles ou à des contraintes structurales.
D’autre part, la présence d’un atome de fluor adjacent à un groupement donneur de liaisons hydrogènes exacerbe l’acidité du proton, augmentant ainsi son caractère donneur.9
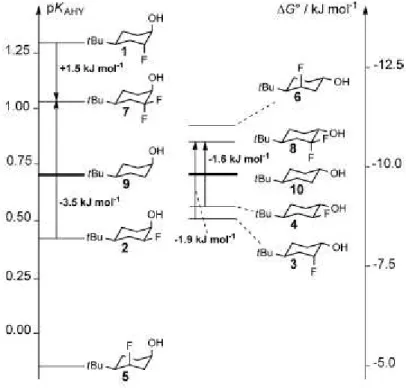
À l’inverse, l’effet inductif attracteur du fluor sur un accepteur proche diminue la densité élec-tronique de celui-ci et donc son aptitude à former des liaisons hydrogènes. Récemment, les travaux de Bruno Linclau ont permis de démontrer que l’effet du fluor sur l’acidité de fonc-tions proches est variable.10 L’introduction d’un ou deux atomes de fluor proche(s) d’une
fonction alcool voisine dans un système cyclique de conformation contrainte, peut diminuer ou augmenter l’acidité de cette fonction (Figure 5). Ceci est dû à la possibilité de former (ou non) une liaison hydrogène intramoléculaire F···H-O permettant de compenser (ou non) l’effet inductif du fluor. Celui-ci est maximal lorsque l’angle dièdre de la fluorhydrine est de 180°, il est réduit de moitié quand l’angle dièdre est de 60°.
7 A. Sun, D. C. Lankin, K. Hardcastle, J. P. Snyder, Chem. Eur. J., 2005, 11, 1579. 8
H.J. Schneider, chemical science, 2012, 3, 1381.
9
J. A. Erickson, J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626.
10 J. Graton, Z. Wang, A. M. Brossard, D. G. Monteiro, J. Y. Le Questel, B. Linclau, Angew. Chem. Int. Ed., 2012,
Figure 5 : Visualisation de l’acidité de fonctions alcools et des différences énergé-tiques entre quelques fluorhydrines.
(Extrait de la publication).10
1.2.4 Influence de la p se e d’ato es de fluor sur la stabilité des
intermé-diaires réactionnels.
1.2.4.1 Radicaux
La stabilité des radicaux en présence d’un atome de fluor est soumise à deux effets : l’effet inductif attracteur du fluor déstabilise les radicaux alors qu’ils sont stabilisés par effet de résonance.11
Lorsqu’un atome de fluor est situé en position α du radical, la structure plane du radi-cal permet un bon recouvrement orbitalaire. L’effet de résonance prédomine permettant de stabiliser le radical (Figure 6). C’est encore le cas avec deux atomes de fluor en position α bien que la structure devienne plus tétraédrique. En ce qui concerne le radical CF3˙ l’effet
inductif prédomine, le radical est alors déstabilisé.
F F F CH3• plan F F F CF3• tétraédrique
Figure 6 : Géométrie de radicaux fluorés.
Dans le cas d’un atome de fluor localisé en position du radical, seul l’effet inductif attracteur influence la stabilité de celui-ci. Les radicaux -fluorés sont donc systématique-ment moins stables que les radicaux non fluorés correspondants (Tableau 6).
Tableau 6 : Énergie de dissociation de liaison C-H (EDL C-H).
CH3-H CH2F-H CHF2-H CF3-H CH3CH2-H CF3CH2-H
EDL C-H
(kcal.mol-1) 104,8 101,2 103,2 106,7 101,1 106,7
1.2.4.2 Carbocations
Comme dans le cas des radicaux, le fluor est capable de stabiliser les carbocations en α par rétrodonation des électrons π (Figure 7), tandis que son effet inductif déstabilise ce
même carbocation.12
C F C F
Figure 7 : Stabilisation par rétrodonation π.
Les carbocations sont davantage stabilisés par un groupement alkyle que par un atome de fluor, il en résulte l’ordre de stabilité suivant en phase gazeuse :13
+CH
3 < +CF3 < +CH2F < +CF2H ≈ +CH2CH3 << +CF2CH3 ≈ +CHFCH3
Les ions -fluoroniums ne sont pas stables, néanmoins leur formation est plus favo-rable que celle d’un ion fluoronium ponté. Ils peuvent se réarranger via des migrations d’alcanes, d’hydrure ou de fluorures (Figure 8).12
12
A. D. Allen, T. T. Tidwell, Advances in carbocation chemistry, 1989, X. Creary Ed., JAI PRESS INC. Greenwich.
13 J. P. Bégué, D. Bonnet-Delpon, Chimie bioorganique et médicinale du fluor, 2005, EDP Sciences/CNRS
F F H F H H H F F F F F F HH 0,0kcal.mol-1 2,82kcal.mol-1 -17,84 kcal.mol-1
Figure 8 : Énergie relative des ions fluoroniums.
La stabilité et la réactivité des ions fluoroniums sera traitée plus en détails dans le chapitre 5.
1.2.4.3 Carbanions
La présence d’un atome de fluor, de chlore ou de brome en position α d’un carbanion permet de stabiliser celui-ci. Étonnamment cette stabilisation est inversement proportionnelle à l’électronégativité de ces halogènes. Ceci peut s’expliquer par un effet de répulsion avec les doublets non liants du fluor qui déstabilise davantage le carbanion fluoré que ceux substi-tués par d’autres halogènes, et ce dû à une plus grande proximité (Figure 9 A).
Les -fluorocarbanions bénéficient à la fois de la stabilisation par effet inductif et par hyperconjugaison (Figure 9 B).14
X
F
A
B
<
Figure 9 : I te a tio de l’ato e de fluo a e les a a io s.
1.2.5 Influence de la p se e d’u ato e de fluor sur le pKa
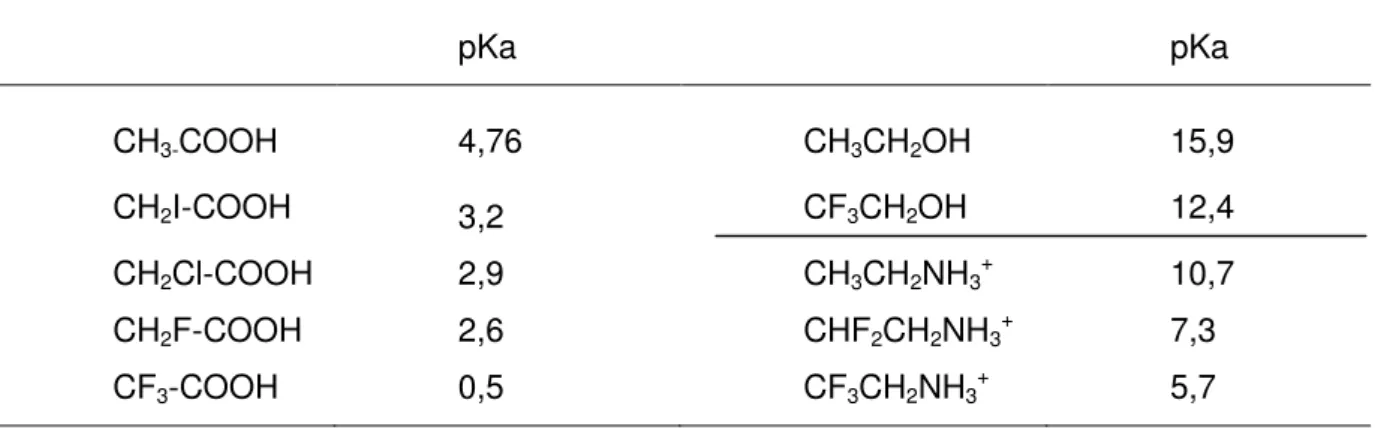
Le pKa étant directement lié à la stabilité de l’anion qui en résulte, on retrouve natu-rellement les propriétés citées ci-avant. Ainsi l’acidité du fluoroforme (pKa = 30,5) est infé-rieure à celle du chloroforme (pKa = 24,4), elle-même inféinfé-rieure à celle du bromoforme (pKa = 22,7).13
À contrario, dans le cas d’hydrogènes non géminaux, l’effet inductif du fluor entraîne une plus forte augmentation de l’acidité que les autres halogènes. L’acidité de fonctions proches telles que les alcools, les acides, les imides est également exacerbée alors que la
14 M. Hudlicky, A. E. Pavlath, Chemistry of Organic Fluorine Compounds II, 1995, ACS Monograph 187,
basicité des amines diminue fortement.15 L’augmentation du nombre d’atomes de fluor
ac-centue sensiblement cet effet (Tableau 7).
Tableau 7 : Valeur de pKa de composés fluorés et non fluorés.
pKa pKa CH3-COOH 4,76 CH3CH2OH 15,9 CH2I-COOH 3,2 CF3CH2OH 12,4 CH2Cl-COOH 2,9 CH3CH2NH3+ 10,7 CH2F-COOH 2,6 CHF2CH2NH3+ 7,3 CF3-COOH 0,5 CF3CH2NH3+ 5,7
1.2.6 Lipophilie
Le caractère lipophile d’un composé est relié au logarithme du coefficient de partage, log P, de celui-ci entre l’eau et l’octanol. L’effet de la fluoration sur la lipophilie dépend forte-ment de la position et du nombre d’atomes de fluor.14
Lorsque le fluor est situé en position α d’un système π ou d’hétéroatomes, hormis les
carbonyles, la lipophilie est généralement augmentée. Cet effet s’estompe lorsque la dis-tance entre l’hétéroatome et le fluor augmente, et au-delà de trois liaisons C-C la lipophilie du composé fluoré est inférieure à celle de son homologue non fluoré (Tableau 8 A).
La substitution d’un hydrogène par un atome de fluor ou par un groupement CF3d’un
composé alcane saturé entraîne une diminution de la lipophilie de ce dernier.
Le cas des composés perfluorés est particulier, leur faible solubilité dans les hydro-carbures étant toutefois supérieure à leur solubilité dans l’eau, le log P en résultant est assez élevé. En réalité ces composés ni lipophiles, ni hydrophiles, forment une troisième phase (Tableau 8 B).
15 M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H.
Tableau 8 : Logarithme du coefficient de partage de composés fluorés et de leurs
homologues non fluorés.
A B
CH3(CH2)nOH Log P CF3(CH2)nOH Log P Composé Log P
n = 1 -0,32 n = 1 0,36 CH4 1,09
n = 2 0,34 n = 2 0,39 CH3F 0,51
n = 3 0,88 n = 3 0,90 CH2F2 0,20
n = 4 1,40 n = 4 1,15 CHF3 0,64
n = 5 1,64 n = 5 1,36 CF4 1,18
2.
Fluor et chimie médicinale
La chimie médicinale s’inspire largement de la nature pour le développement de nou-velles substances actives. Après la découverte d’un composé d’intérêt biologique naturel, celui-ci est modulé afin d’accéder à une chimiothèque pouvant permettre l’émergence de principes actifs plus efficaces. Le fluor est extrêmement rare dans les composés organiques naturels, seules quelques plantes et deux bactéries produisent des composés fluorés.16
Ain-si, la chimie médicinale est longtemps restée éloignée de cet élément.
Les premières substances actives fluorées n’apparaissent que dans les années 1950, dès lors la chimie du fluor a connu un développement extrêmement rapide. Depuis 1970 la part de marché des composés fluorés dans le domaine pharmaceutique est passée de 3 à 25%. Dans le domaine agrochimique ils représentent aujourd’hui près de 50% du marché.2
2.1 Intérêt des motifs fluorés en chimie thérapeutique.
2.1.1 Biodisponibilité
L’introduction d’un atome de fluor sur un composé organique peut permettre d’augmenter sa lipophilie, or cette propriété est d’une importance capitale en chimie
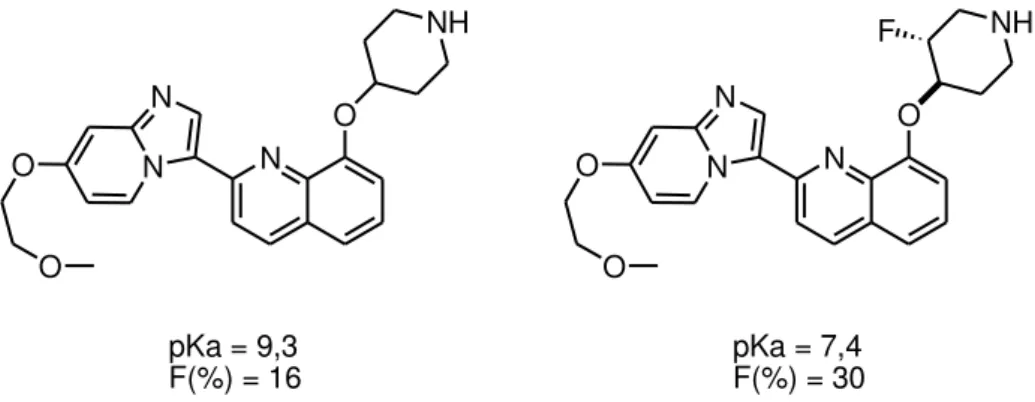
nale. L’augmentation de la lipophilie permet un meilleur passage membranaire et donc une meilleure biodisponibilité. Ce gain en termes de biodisponibilité est encore accru dans le cas des composés azotés. En effet la présence d’un atome de fluor à proximité de la fonction azotée permet une diminution de son caractère basique. Cet effet est particulièrement impor-tant lorsqu’il s’agit de fonctions protonées à pH physiologique telles que les amines. Par exemple, la substitution d’un hydrogène par un atome de fluor dans les molécules de type imidazopyridine présentant une activité anticancéreuse, a permis d’augmenter efficacement la biodisponibilité de ces composés (Figure 10).17 Cette biodisponibilité est exprimée en
pourcentage de substance active présente dans le réseau sanguin par rapport à la quantité de substance active administrée.
N N O O N O NH N N O O N O NH F pKa = 9,3 F(%) = 16 pKa = 7,4F(%) = 30
Figure 10 : pKa et biodisponibilité (F) d’inhibiteurs de récepteurs de facteur de crois-sance plaquettaire.
2.1.2 Résistance métabolique
La présence d’un atome de fluor ou d’un groupement fluoré peut parfois améliorer la stabilité métabolique d’un composé. L’augmentation du temps de demi-vie d’une substance active permet de limiter les dosages d’un traitement et la fréquence d’administration. Dans ce contexte, le fluor est couramment utilisé afin de bloquer le métabolisme oxydatif (Figure 11).18
Par exemple, dans le cas de composés dérivés d’analgésiques muscariniques utilisés dans la lutte contre le syndrome du côlon irritable, la présence du groupement trifluoromé-thyle inhibe l’oxydation métabolique du composé. La demi-vie du composé est alors considé-rablement augmentée, en effet une heure après administration à des rats, la concentration
17 E. J. Hicken, F. P. Marmsater, M. C. Munson, S. T. Schlachter, J. E. Robinson, S. Allen, L. E. Burgess, R. K.
DeLisle, J. P. Rizzi, G. T. Topalov, Q. Zhao, J. M. Hicks, N. C. Kallan, E. Tarlton, A. Allen, M. Callejo, A. Cox, S. Ra-na, N. Klopfenstein, R. Woessner, J. P. Lyssikatos, ACS Med. Chem. Lett., 2014, 5, 78.
18
C. H. Mitch, T. J. Brown, F. P. Bymaster, D. O. Calligaro, D. Dieckman, L. Merrit, S. C. Peters, S. J. Quimby, H. E. Shannon, L. A. Shipley, J. S. Ward, K. Hansen, P. H. Olesen, P. Sauerberg, M. J. Sheardown, M. D. B. Swedberg, P. Suzdak, B. Greenwood, J. Med. Chem., 1997, 40, 538.
plasmatique du dérivé trifluorométhylé est 40 fois supérieure à celle de son homologue non fluoré. N N S N S N N S N S F F F 21* 805*
Figure 11 : Analgésique muscarinique et son homologue fluoré
*concentration plasmatique, 1heure après administration de 30 mg.kg-1 (ng.mL-1).
2.1.3 Biomimétisme
Une des stratégies utilisée en chimie médicinale consiste en la conception de bio-mimes. Le remplacement d’une fonction chimique d’un substrat enzymatique par un grou-pement équivalent en volume et/ou en propriétés stéréoélectroniques permet la reconnais-sance du mime par l’enzyme. Les propriétés physico-chimiques du fluor font de lui un bon bioisostère des atomes d’oxygène et d’hydrogène. De nombreux mimes fluorés de sucres et de peptides ont été utilisés comme inhibiteurs d’enzymes ou comme outils d’investigations dans l’étude des mécanismes enzymatiques.19 Par exemple, cette méthode a été utilisée
dans le cas de l’étude mécanistique de la réaction de déshydratation biosynthétique de nu-cléoside-diphosphate-glucose (NDP-glucose), catalysée par l’enzyme glucose-4,6-déshydratase (Figure 12). O HO H OH HO HO ONDP O OH HO HO ONDP N N H H CONH2 R R CONH2 O HO HO ONDP O O HO HO ONDP O Eod R = ADP-ribosyl O
Figure 12 : Cycle catalytique de la glucose-4,6-déshydratase.
Le passage par un transfert d’hydrure a pu être mis en évidence grâce à l’utilisation d’un mime fluoré de cytidine-diphosphate-glucose (CDP-glucose) (Figure 13). L’élimination d’un ion fluorure entraine la formation d’une liaison covalente entre l’enzyme et le substrat. Ce complexe a été caractérisé par spectroscopie de masse, le départ d’ion fluorure par RMN
19F. O O H CHF 2 HO HO OCDP O HO OHOCDP O O HO HO OCDP O Eod NAD H B H F F B NAD H F H NAD H BH NAD O HO HO OCDP O F HH BH NAD O HO HO OCDP O H H BH Nu O HO HO OCDP NAD B O Nu
Figure 13 : Étude mécanistique via l’utilisation de mime fluoré.
Parmi les différents composés fluorés, nous nous sommes plus particulièrement inté-ressés aux dérivés -fluoroazotés. Le chapitre suivant est consacré aux méthodes permet-tant leur accès.
2.2 Méthodes de synthèse de
ȕ-fluoroamines
Depuis le milieu des années 1950, la chimie du fluor a connu un essor considérable. Les composés fluorés se retrouvent dans tous les domaines de la chimie et leur nombre est en constante augmentation. Les méthodes pour accéder à ces motifs ont évolué en consé-quence. Aujourd’hui le chimiste organicien dispose d’un véritable arsenal synthétique20,13,14,21
20
T. Liang, C. N. Neumann, T. Ritter, Angew. Chem. Int. Ed., 2013, 52, 8214.
21 I. Ojima, J. R. McCarthy, J. T. Welch, Biomedical Frontiers Fluorine Chemistry, 1996, A.C.S. SYMPOSIUM
permettant l’obtention de composés fluorés de plus en plus complexes. Les méthodes de fluoration décrites ci-après représentent les principales stratégies utilisées aujourd’hui pour l’obtention de -fluoroamines sans en dresser la liste exhaustive.
2.2.1 Fluoration par voie nucléophile
2.2.1.1 Substitution
nucléophile d’amino-alcool
La stratégie de synthèse la plus communément utilisée passe par l’utilisation du Xtal-Fluor-E® DAST ou du Deoxofluor® (Figure 14). Ces réactifs commerciaux et faciles à
manipu-ler permettent la substitution nucléophile d’alcool ou de carbonyle par respectivement un ou deux atomes de fluor.
N S F F F DAST N S F F F O O Deoxofluor N S F F XtalFluor-E
Figure 14 : Structure des principaux agents de fluoration nucélophile.
Cette substitution induit une inversion de configuration dans le cas des alcools per-mettant un bon contrôle du centre stéréogène. Malheureusement dans le cas d’amino alcool cette méthodologie conduit souvent des réarrangements22 dus au passage par un
intermé-diaire ion aziridinium (Figure 15) ou des éliminations.23 De plus, elle est sensible à
l’encombrement stérique ainsi qu’à la participation de fonctions proches de l’hydroxyle.24
N R' R'' OH DAST N R' R'' O S N F F F N R' R'' F R R R N R' R'' F R N R' R R'' F H N R' R'' R thermodynamique cinétique
Figure 15 : Mécanisme de fluoration par le DAST
22 (a) B. Duthion, D. G. Pardo, J. Cossy, Org. Lett., 2010, 12, 4620.
(b) B. Drouillat, F. Couty, O. David, G. Evano, J. Marrot, Synlett, 2008, 9, 1345.
23
J. L. Castro, I. Collins, M. G. N. Russel, A. P. Watt, B. Sohal, D. Rathbone, M. S. Beer, J. A. Stanton, J. Med.
Chem., 1998, 41, 2667.
2.2.1.2
Fluoration d’alcool benzylique
L’accès aux -fluoroamines est également possible par substitution nucléophile, par du fluorure de césium, d’un alcool benzylique tosylé. La formation d’un carbanion en pré-sence de diisopropylamidure de lithium permet son addition sur une imine tosylée et d’accéder au motif souhaité (Figure 16).25
SOp-Tol O Ts CsF [mim-tOH][OMs] S F OH p-Tol S F O p-Tol Li LDA R N R' SOp-Tol SOp-Tol F HN SOp-Tol R' R tBuLi F H2N R' R
Figure 16 : Voie de s th se pou la fluo atio d’al ools e zili ues.
2.2.1.3 O
uverture d’aziridine
Une autre approche consiste en la synthèse d’aziridine26 puis ouverture de celle-ci
par un agent de fluoration nucléophile. L’agent fluorant est généralement un complexe HF·base27 mais des réactifs donneurs d’ions fluorure peuvent également être utilisés dans le
cas d’aziridines bicycliques28.
Cette méthode est efficace pour l‘obtention de -fluoroamines, mais la régiosélectivité et la stéréosélectivité de la réaction dépendent de la stabilité de l’intermédiaire ion aziridi-nium formé après protonation ainsi que des ions carbéaziridi-niums potentiellement mis en jeu (Figure 17). R1 R3 R2 R4 N R R1 R3 R2 R4 N R H R1 R3 R2 R4 NH R R2 R3 R1 R4 NH R F R3 R4 NH R F R1 R2 R3 R4 NH R F R2 R1 F R1 R2 NH R F R3 R4 R1 R2 NH R F R4 R3 R1 R2 NH R R3 R4 R1 R2 NH R R4 R3
Figure 17 : Mécanisme de fluo atio d’azi idi es.
25 J. L. Garcia Ruano, A. Parra, I. Alonso, S. Fustero, C. Del Pozo, Y. Arroyo, A. Sanz-Tejedor, Chem. Eur. J., 2011,
17, 6142.
26
J. A. Kalow, D. E. Schmitt, A. G. Doyle, J. Org. Chem., 2012, 77, 4177.
27 G. Alvernhe, A. Laurent, G. Haufe, J. Fluorine Chem., 1986, 34, 147.
2.2.1.4 S
ubstitution d’halogénoamine
Les -fluoroamines peuvent également être synthétisées par substitution d’une halo-génoamine avec des fluorure d’ammoniums ou de potassium. La synthèse de ces halogé-noamines est généralement réalisée par activation d’un -aminoalcool, puis substitution par un ion halogénure. Malheureusement l’alcool activé peut subir une attaque intramoléculaire de l’amine pour former un ion aziridinium, ouvert ensuite par l’ion halogénure (Figure 18).29
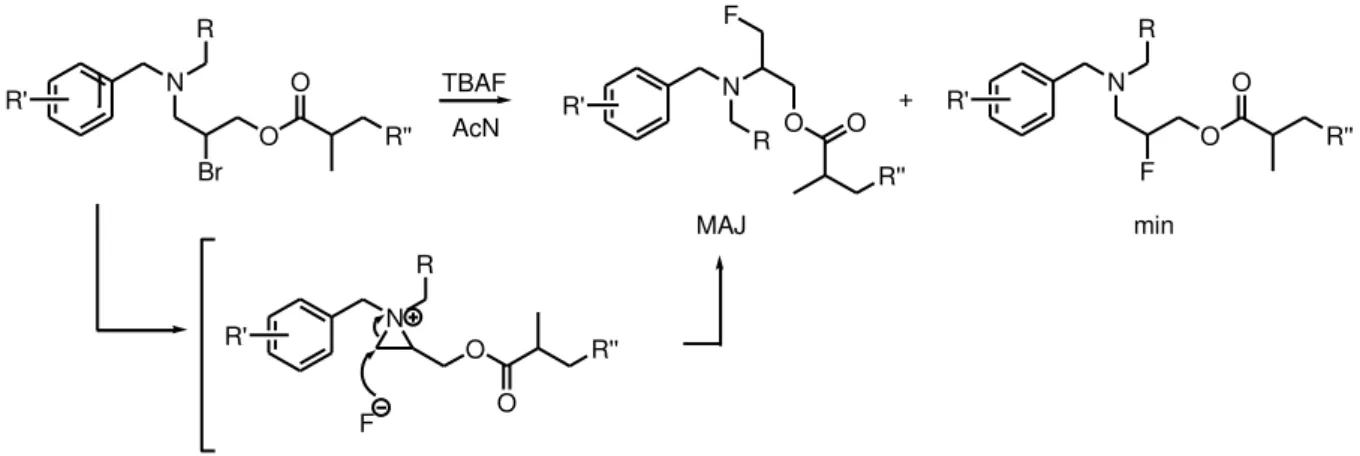
Cette voie de synthèse souffre d’un manque de régiosélectivité et de stéréosélectivité comme dans le cas de l’ouverture d’aziridines précédemment évoqué (Figure 19).30
Malgré ces difficultés, cette synthèse est encore utilisée notamment dans le domaine de l’imagerie médicale. Le fluor 18, utilisé en tomographie par émission de positrons, a un temps de demi-vie de 110 minutes. L’échange halogène-fluor pouvant être réalisé en 15 mi-nutes, cette méthode reste un bon moyen d’obtenir les composés désirés en un minimum de temps29. OH N R O N R N R Cl N R Cl N R MsCl S O O Cl Cl +
Figure 18 : Mécanisme de réarrangement lors de la synthèse d’halogénoamine.
R' N R Br O O R'' R' N R R' N R F O O R'' F O O R'' + TBAF AcN R' N R O O R'' F MAJ min
Figure 19 : Mécanisme de réarrangement lors de la fluo atio d’halog oa i e.
29
S. Nag, G. Kettschau, T. Heinrich, A. Varrone, L. Lehmann, B. Gulyas, A. Thiele, É. Keller, C. Halldin, Bioorg.
Med. Chem., 2013, 186.
2.2.2 Fluoration par voie électrophile
La fluoration par voie électrophile a été largement étudiée. De nombreux réactifs ont été développés pour réaliser ce type de réaction.31 Les composés de type
N-fluorosulfinimides et ammoniums fluorés sont principalement utilisés (Figure 20).
S N S F O O O O NFSI N N Cl F BF4 BF4 Selectfluor N F Tf Triflate de pyridinium
Figure 20 : Structures des principaux agents de fluoration électrophile.
2.2.2.1
α-fluoration d’imines et réduction
La synthèse de -fluoroamines est généralement réalisée en trois étapes, fluoration, condensation d’une amine sur un carbonyle et réduction (Figure 21).32L’ordre de ces étapes
peut varier.33 Ph H O NFSI Ph H O F HN N Boc Ph H N F N Boc Ph H N F N Boc NaBH(OAc)3
Figure 21 : S th se d’a i es fluo es pa oie le t ophile.
2.2.3 Aminofluoration
2.2.3.1 Fluoration catalysée par le fer
Très récemment une nouvelle voie de synthèse catalysée par du fer II en présence d’ions fluorure a été proposée.34 Les auteurs proposent un mécanisme de type radicalaire
(Figure 22).
31
G. Sankar Lal, G. P. Pez, R. G. Syvret, Chem. Rev., 1996, 96, 1737.
32
O. O. Fadeyl, C. W. Lindsley, Org. Lett., 2009, 11, 943.
33 G. Verniest, E. Van Hende, R. Surmont, N. De Kimpe, Org. Lett., 2006, 8, 4767. 34 D. F. Lu, G. S. Liu, C. L. Zhu, B. Yuan, H. Xu, Org. Lett., 2014, 16, 2912.
Ph O O N FeLn RO X Ph O O HN OR Ph O O N FeLn F F N O O Ph H FeLn F F • N H O O Ph F FeX2Ln -HX Et3N•3HF XtalFluor-E HX
Figure 22 : Voie de s th se pou l’a i ofluo atio atal s e pa le fe .
2.2.3.2 Aminofluoration de styrène palladocatalysée
Une aminofluoration directe de composés insaturés, catalysée par le palladium a été proposée (Figure 23). Cette synthèse, bien qu’efficace, est limitée à la famille des styrènes.35
R + NFSI Pd(OAc)2
R
N(SO2Ph)2 F
Figure 23 : Aminofluoration catalysée par le palladium.
2.2.4 Utilisation de synthons fluorés
Une alternative possible consiste en la préparation de synthons fluorés puis introduc-tion du dérivé aminé.
2.2.4.1 Fluoroalcools
Les -fluoroalcools peuvent être utilisés comme synthons. La stratégie de synthèse repose sur une tosylation de l’hydroxyle, puis substitution de celui-ci par une amine permet-tant l’accès aux -fluoroamines correspondantes.36 Les températures élevées nécessaires à
cette synthèse limitent son utilisation et entraînent des éliminations (Figure 24).
F OTs H N DMSO 150°C F N + OTs +
Figure 24 : Su stitutio d’al ool tos l pa la diméthylamine.
2.2.4.2 Fluoroalkyles
L’introduction d’un groupement fluorométhyle sur un composé aminé a également été étudiée. Les réactifs de fluoroalkylation utilisés peuvent être de type nucléophile, électrophile
35 S. Qiu, T. Xu, J. Zhou, Y. Guo, G. Liu, J. Am. Chem. Soc., 2010, 132, 2856. 36 C. Toulgui, M. M. Chaabouni, A. Baklouti, J. Fluorine Chem., 1990, 46, 385.
ou radicalaire.37Dans l’exemple qui suit, le fluoro-bis(phénylsulfonyl)méthane est déprotoné
par le n-BuLi, le fluorocarbanion correspondant peut alors réagir par addition nucléophile sur l’imine conduisant au dérivé fluoro-bis(phénylsulfonyl)méthylé. Une désulfonylation réduc-trice permet l’obtention du composé -fluoroamine (Figure 25).38
Ph N (PhSO 2)2CHF Ph CF(SO2Ph)2 NH Mg MeOH/HCl Ph CH2F NH2 Ph N FC(SO2Ph)2 n-BuLi n-BuLi S t-Bu O S O t-Bu S t-Bu O
Figure 25 : Mécanisme de fluorométhylation nucléophile.
2.2.4.3 Fluorohalogénés
Une réaction d’halofluoration de composés insaturés permet l’accès à des synthons fluorés fonctionnalisables. Généralement ces synthons sont préparés par halogénation élec-trophile à l’aide de NXS puis fluoration par un complexe HF/base.39 La substitution
nucléo-phile de l’halogène par une amine ou un précurseur d’amine, permet l’obtention du composé -fluoroamine désiré (Figure 26).
TrN N NBS/HF•Et3N TrN N F Br TrN N F Phtal N O O K NH2NH2 TrN N F H2N
Figure 26 : Voie de s th se pa su stitutio d’u o pos fluo ohalog
pa u p
u seu d’a i e.
37
J. Hu, W. Zhang, F. Wang, Chem. Commun., 2009, 7465.
38 J. Liu, L. Zhang, J. Hu, Org. Lett., 2008, 10, 5377. 39 B. Dolensky, K. L. Kirk, J. Org. Chem., 2001, 66, 4687.
2.2.5 Fluoration en milieu superacide
Une méthode de fluoration directe de composés azotés insaturés en milieu supera-cide a été développée au laboratoire. Le dication ammonium-carbénium, obtenu après pro-tonation de l’azote puis de la double liaison, a un caractère superélectrophile (*espèce ca-pable de réagir avec des nucléophiles faibles). Celui-ci peut alors être piégé par les ions fluo-rure du milieu permettant l’accès aux -fluoroamines (Figure 27).40
R' N R R' N R H R' N R H R' N R H F H+ H+ H+
Figure 27 : Hydrofluoration par activation superélectrophile.
L’utilisation de conditions superacides permet de protoner la plupart des substances organiques. La réactivité des substrats dans ces milieux est donc très différente de celle en milieu plus « conventionnel ». Les caractéristiques des milieux superacides sont dévelop-pées dans la partie suivante.
3.
Les milieux superacides
L’acide est connu depuis l’antiquité, ce terme est initialement lié aux propriétés gusta-tives des composés. Le terme superacide est bien plus récent, ce n’est qu’en 1927 que Co-nant et Hall l’utilisent pour la première fois.41 Il est repris au début des années 1960 par le Pr
Georges Olah qui utilise ces milieux très ionisants pour l’étude de carbocations. Celui-ci re-çut le prix Nobel de chimie en 1994 pour ces travaux.
Le milieu superacide a été défini par Gillespie au début des années 1960 comme étant un milieu plus acide que l’acide sulfurique pur pour les acides de Brønsted. Olah a en-suite proposé le chlorure d’aluminium comme référence arbitraire pour les acides de Lewis,42
tout acide de Lewis plus acide que AlCl3 est alors considéré comme un superacide de Lewis.
Depuis, l’utilisation des superacides s’est largement développée, notamment dans le domaine de la pétrochimie pour la valorisation de paraffines. Ils peuvent également être utili-sés pour la transformation de molécules plus complexes, c’est le cas notamment pour la
40 S. Thibaudeau, A. Martin-Mingot, M. P. Jouannetaud, O. Karam, F. Zunino, Chem. Commun., 2007, 3198.
(* Cette notion de superélectrophile sera développée par la suite.
41
N. F. Hall, J. B. Conant, J. Am. Chem. Soc., 1927, 49, 3047.
42 G. A. Olah, G. K. S. Prakash, A. Molnar, J. Sommer, Superacid Chemistry, 2009, 2nd Edition John Wiley and
thèse de la vinflunine, mise au point au laboratoire par Jacquesy43et aujourd’hui
commercia-lisée par Pierre Fabre sous le nom Javlor®.
Les superacides existent aujourd’hui sous forme liquide ou solide (supporté sur char-bon, sur oxyde métallique ou de type zéolithe), constitué d’acide de Brønsted, d’acide de Lewis, de mélange binaire ou ternaire. L’acidité peut varier énormément selon la composition du milieu, impliquant une réactivité différente des substrats.
3.1 Propriétés des milieux superacides
3.1.1 Mesu e de l’a idit
Les milieux superacides étant anhydres, la mesure de l’acidité nécessite l’utilisation de bases faibles. Dans ces milieux extrêmes, ces bases faibles s’ionisent par protonation, permettant le calcul de la fonction d’acidité de Hammet (H0) des différents superacides.42
Cette valeur est définie par l’équation (1).
H0 = pKBH+ - log BH +
B
(1)
Les concentrations en espèces ionisées ou non ionisées peuvent être déterminées par plusieurs méthodes. L’une des techniques les plus utilisées est la spectroscopie UV-Visible de dérivés présentant une longueur d’onde différente au maximum d’absorption sous forme ionisé et sous forme neutre. H0 est dépendant de la base utilisée sauf pour une série
de composés appelés bases de Hammet tel que les anilines ou les nitrobenzènes qui seront donc utilisés comme référence pour les mesures de H0. Les milieux superacides sont plus
acides que l’acide sulfurique pur dont la valeur de H0 définie arbitrairement est de -12. Ainsi
certains acides de Brønsted sont des superacides, tels que l’acide triflique ou l’acide fluorhy-drique dont les valeurs de H0 sont respectivement de -14,1 et -15,1.
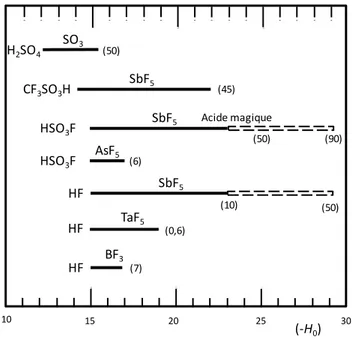
3.1.2 Différents types de superacides
Les superacides de Brønsted peuvent être associés à un superacide de Lewis afin d’atteindre des acidités supérieures. Les mélanges les plus utilisés sont binaires, l’acidité du milieu dépend alors de la proportion d’acide de Lewis ajoutée (Figure 28). Des mélanges ternaires ont également été étudiés.
43 J. Fahy, A. Duflos, J. P. Ribet, J. C. Jacquesy, C. Berrier, M. P. Jouannetaud, F. Zunino, J. Am. Chem. Soc., 1997,
Figure 28 : Valeu de la fo tio d’a idit de Ha
et (H
0) de différents mélanges binaires
e fo tio de la p opo tio d’a ide de Le is.
Barre pleine : mesurée expérimentalement en présence d’un indicateur. Barre en pointillés : estimée par mesure cinétique.
Concentration en ( ) mol% acide de Lewis.
Au laboratoire, le milieu superacide généralement utilisé est généré par un mélange d’acide fluorhydrique anhydre et de pentafluorure d’antimoine. La variation de la proportion d’antimoine permet de couvrir une large gamme d’acidité de H0 = -15,1 à H0 ≈ -25. Ce
mé-lange permet d’atteindre l’acidité la plus forte connue à ce jour.
3.1.3 HF anhydre
L’acide fluorhydrique anhydre a une température d’ébullition de 20 °C à pression at-mosphérique. Dans les conditions normales de température et de pression il est donc à l’état gazeux. Une fois condensé, il se présente sous forme de liquide incolore fumant. Composée de chaines polymères de molécules de HF liées entre elles par des liaisons hydrogène forte, la longueur de chaine est en moyenne de sept monomères HF44. Le HF anhydre a une
va-leur de H0 de -15,1 faisant de lui un superacide de Brønsted (Figure 29).
44 S. E. McLain, C. J. Benmore, J. E. Siewenie, J. Urquidi, J. F. C. Turner, Angew. Chem. Int. Ed., 2004, 43, 1952.
10 15 20 25 30 (-H0) SbF5 H2SO4 SO3 Acide magique AsF5 BF3 TaF5 HSO3F HSO3F SbF5 (6) (50) (90) (10) (50) (0,6) (7) (50) CF3SO3H SbF5 (45) HF HF HF
Figure 29 : Structure du HF anhydre modélisé à partir des données obtenues par DRX.
La faible température d’ébullition, la très forte acidité et la toxicité de l’acide fluorhy-drique impliquent de prendre beaucoup de précautions quant à l’utilisation de ce composé.
3.1.4 Composition du mélange HF/SbF
5L’ajout de SbF5 au HF anhydre permet d’accroitre considérablement l’acidité du
mi-lieu. L’ajout de SbF5 conduit à une dissociation des molécules de HF, entrainant la formation
de complexes anioniques SbF6- et d’espèces H3F2+. Lorsque la proportion en SbF5
aug-mente, les complexes anioniques d’antimoine conduisent à des espèces polymériques de type SbnF5n+1- (Figure 30).45 Parallèlement les cations H3F2+ sont remplacés par des ions
H2F+. La diminution de la solvatation des ions H+ entraîne une augmentation de l’acidité46.
Figure 30 : Espèces présentent dans le milieu en fonction de la proportion de SbF
5.
45 J. C. Culmann, M. Fauconet, R. Jost, J. Sommer, New J. Chem., 1999, 23, 863. 46 P. M. Esteves, A. Ramirez-Solis, C. J. A. Mota, J. Am. Chem. Soc., 2002, 124, 2672.
L’augmentation de la taille des anions entraîne une meilleure délocalisation de la charge négative, l’ajout de SbF5 diminue donc le caractère fluorant du milieu (Figure 31).
Figure 31 : Structure des anions de type Sb
nF
5n+1-.
Dans ces milieux d’extrême d’acidité il est possible de générer des espèces superé-lectrophiles, cette notion de superélectrophilie sera développée ci-après.
4.
Les superélectrophiles
Les carbocations sont connus depuis plus d’un siècle,47 mais ce n’est qu’au début
des années 1960 qu’ils sont observés par Olah. Cette étude a été rendue possible par l’utilisation des superacides, en effet dans ces milieux très ionisants, les carbocations ont une durée de vie accrue. Dans ces conditions il est possible de former des espèces polyca-tioniques par polyprotonation.
La répulsion des charges induit une déstabilisation de ces polycations entrainant une augmentation de leur réactivité. La position relative des charges joue un rôle primordial sur l’électrophilie de ces composés. Lorsque les charges sont suffisamment proches les espèces chargées sont alors capables de réagir avec des nucléophiles très faibles tels que les ions fluorure solvatés dans le mélange HF/SbF5, ou des aromatiques non activés voire même
désactivés (Figure 32).48 Ces espèces extrêmement réactives sont alors appelées
superélec-trophiles. CH3 NH2 Ph OH Ph H+ CH3 NH3 Ph Ph C6H6 CH3 NH2 Ph Ph Ph
Figure 32 : Superélectrophile dicationique ammonium-carbénium piégé par du benzène.
47 G. A. Olah, D. A. Klumpp, Superelectrophiles and their Chemistry, 2008, John Wiley and Sons Inc., Hoboken. 48 D. A. Klumpp, S. L. Aguirre, G. V. Sanchez Jr., S. J. De Leon, Org. Lett., 2001, 3, 2781.
-4.1 Superélectrophiles gitoniques et distoniques
La réactivité des superélectrophiles dépend fortement de la distance entre les charges.
Les intermédiaires de type gitonique sont porteurs d’au moins deux charges distantes de moins de deux atomes. Lorsque les charges sont portées par un même atome le superé-lectrophile gitonique est dit geminal. Les deux charges peuvent être portées par deux atomes voisins il s’agit alors d’un superélectrophile gitonique vicinal. Les 1,3-dications sont également considérés comme gitoniques.
Lorsque les charges sont distantes de deux atomes ou plus ils sont appelés
disto-niques. Les charges peuvent être portées indépendamment par un atome de carbone ou un
hétéroatome. Quelques exemples de ces différents superélectrophiles sont reportés ci-après (Tableau 9). Par la suite, seul sera développé le cas des superélectrophiles azotés.
Tableau 9 : Exemples de superélectrophiles dicationiques.
Superélectrophiles gitoniques
Superélectrophiles distoniques
geminaux
vicinaux
1,3-dications
C H H H H H H 2+ (49) H H H H (50) Ph P Ph Ph (51) OH H X (52) O H H H H 2+ (53) C N H H (54) O (55) (56) 2+ H N H H H H (57) O N O H (58) O O H H (59) N H (60)
49 G. A. Olah, G. K. S. Prakash, G. Rasul, J. Org. Chem., 2001, 66, 2907. 50
K. Lammertsma, M. Barzaghi, G. A. Olah, J. A. Pople, A. J. Kos, P. V. R. Schleyer, J. Am. Chem. Soc., 1983, 105, 5253.
51 Y. Zhang, S. L. Aguirre, D. A. Klumpp, Tetrahedron Lett., 2002, 43, 6837.
52 A. V. Vasilyev, S. Walspurger, P. Pale, J. Sommer, Tetrahedron Lett., 2004, 45, 3379.
53 V. Prakash Reddy, E. Sinn, G. A. Olah, G. K. Surya. Prakash, G. Rasul, J. Phys. Chem. A, 2004, 108, 4036. 54 Y. Sato, M. Yato, T. Ohwada, S. Saito, K. Shudo, J. Am. Chem. Soc.,1995, 117, 3037.
55 D. Farcasiu, G. Miller, S. Sharma, J. Phys. Org. Chem., 1990, 3, 639.
56 A. Meijere, O. Schallner, P. Golitz, W. Weber, P. V. R. Schleyer, G. K. Surya Prakash, G. A. Olah, J. Org. Chem.,
1985, 50, 5255.
57
G. A. Olah, A. Burrichter, G. Rasul, G. K. Surya. Prakash, J. Am. Chem. Soc., 1997, 119, 4594.
58
G. A. Olah, A. Germain, H. C. Lin, D. A. Forsyth, J. Am. Chem. Soc., 1975, 97, 2928.
59 G. A. Olah, N. Hartz, G. Rasul, A. Burrichter, G. K. Surya Prakash, J. Am. Chem. Soc., 1995, 117, 6421. 60 D. A. Klumpp, P. S. Beauchamp, G. V. Sanchez Jr., S. Aguirre, S. De Leon, Tertrahedron Lett., 2001, 42, 5821.
4.2 Superélectrophiles azotés
4.2.1 Réaction de type Friedel Crafts
Dans la littérature, les superélectrophiles azotés sont souvent générés pour réaliser des réactions de type Friedel Crafts, intramoléculaires ou intermoléculaires, avec des aroma-tiques non activés, voire désactivés. Les dérivés azotés utilisés possèdent des groupements nitrile, nitro, amine, aromatique ou hétéroaromatique.
4.2.1.1 Voie intramoléculaire
Les réactions de Friedel Crafts intramoléculaires permettent l’accès à des composés polycycliques. Le superacide utilisé le plus couramment pour réaliser ce type de réaction est l’acide triflique.
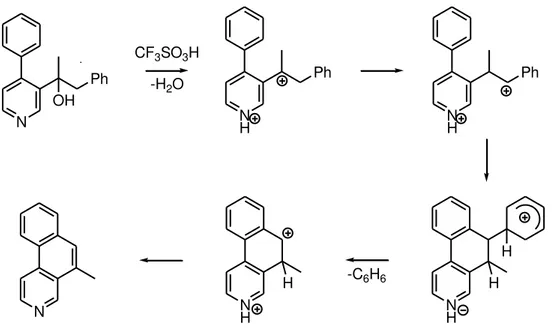
Une réaction de Houben-Hoesch a ainsi été réalisée sur un 4-phénylbutyronitrile en présence d’acide triflique afin d’accéder au composé dihydronaphtalène correspondant (Figure 33).54 Une étude cinétique a été menée ; elle met en évidence le passage par une
espèce dicationique après diprotonation du groupement nitrile.
C N C N H H N CF3SO3H H O H2O
Figure 33 : Réaction de Houben-Hoesch intramoléculaire.
Dans des conditions analogues, Klumpp et son équipe ont réalisé la synthèse de composés polycycliques aromatiques azotés.61En présence d’acide triflique, une
déshydra-tation a lieu. L’isomérisation du cation initialement formé résulte de la répulsion électrosta-tique. Le dication finalement obtenu est piégé par l’aromatique voisin. Après perte d’une mo-lécule de benzène, le composé polycyclique aromatique azoté est obtenu (Figure 34).