JOAHNN HERNANDO PALACIOS RIOS
AMELIORATION D'UN ECHANTILLONNEUR
À FLUX PASSIF POUR LA MESURE
DE L'OXYDE NITREUX (N
20)
Mémoire présenté
à la Faculté des études supérieures de l'Université Laval dans le cadre du programme de maîtrise en Génie Agroalimentaire
pour l'obtention du grade de maître es sciences (M.Sc.)
DEPARTEMENT DES SOLS ET DE GENIE AGROALIMENTAIRE FACULTÉ DES SCIENCES DE L'AGRICULTURE ET DE L'ALIMENTATION
UNIVERSITÉ LAVAL QUÉBEC
2010
Résumé
L'oxyde nitreux (N2O) est l'un des gaz à effet de serre (GES) parmi les plus puissants et il joue un rôle important dans le réchauffement global. Pour mesurer les sources à faibles émissions de N2O, les approches conventionnelles sont souvent sophistiquées et coûteuses et l'approche par échantillonnage à flux passif montre un intérêt. Le but de la présente recherche est d'optimiser la performance de l'échantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005) dans les différentes étapes de son utilisation : le conditionnement de l'appareil, l'adsorption et la désorption du N2O.
L'échantillonneur de Godbout et al. (2006) et Gaudet (2005) utilise une couche de 2 mm de zéolite pour adsorber le N2O. Lorsque l'air passe à travers l'échantillonneur à une vitesse proportionnelle à la vitesse de l'air à l'extérieur, la zéolite adsorbe le N2O. Par la suite, le N2O contenu dans l'adsorbant est désorbé thermiquement. La concentration de gaz est déterminée à l'aide d'un chromatographic à gaz et ensuite, la masse désorbée est calculée afin d'obtenir la valeur de l'émission de N2O.
Dans le cadre de cette étude, l'échantillonneur a été étanchéifié et modifié pour en faciliter l'assemblage. L'épaisseur de l'adsorbant a été augmentée à 4, 6 et 8 mm, ce qui a permis d'augmenter sa capacité d'adsorption de 96, 187 et 275 % respectivement. Les résultats de l'évaluation de la performance de l'échantillonneur modifié montrent une augmentation de l'efficacité d'adsorption de 91 % (Gaudet 2005) à 96,5, 98,3, 98,3 et 98,4 % et une augmentation de l'efficacité de désorption de 68 % à 83 %, de 57 % à 75 %, de 45 % à 82 % et de 30 % à 74 % pour les échantillonnées pourvus de couches de 2, 4, 6 et 8 mm respectivement. Également, la conception des montages et des modes opératoires ont permis à l'échantillonneur d'obtenir des mesures avec une exactitude moyenne de 77,3 % et une variabilité moyenne de 13,6 %. Suite aux résultats, il est possible d'affirmer que les modifications apportées à l'échantillonneur et aux procédures d'analyse ont permis d'améliorer ses performances et de faire progresser son développement.
Remerciements
Je tiens en premier lieu à remercier mes directeurs de recherche pour leurs judicieux conseils et leur confiance. Ils ont fait de ce travail une véritable expérience de croissance professionnelle et personnelle.
Mes remerciements s'adressent aussi à tous les membres de l'équipe Mésanges MD de
l'Institut de recherche et de développement en agroenvironnement (IRDA) ainsi qu'à Christian Gauthier, ouvrier (IRDA), qui ont collaboré pour le progrès de cette recherche en faisant toujours preuve de professionnalisme.
Je tiens également à exprimer toute ma gratitude à ma famille, spécialement à mes parents, ma sœur, mes grands-parents et mon oncle, pour leur fervent amour qui, malgré la distance, m'a tenu en vie tout au long de ce défi.
Un dernier merci, mais non le moindre, à ma nouvelle famille du Québec. Il s'agit de mes amis Miguel, Rônja, Paola, Carolina, Sylvie, Julie et Alexandre que je remercie pour l'encouragement et l'amitié inconditionnelle qu'ils m'ont offerts. Enfin, j'ai une pensée particulière pour mes amis en Colombie, notamment Carlos, Giovanni, Luis Gabriel, Stiward et William.
A mon grand-père, qui est parti mais qui a laissé en moi la trace d'une personne exemplaire à admirer
Table des matières
Résumé ii Remerciements iii
Table des matières v Liste des tableaux vii
Liste des figures ix Introduction 1 1. Mise en contexte 2
1.1 Émission de gaz à effet de serre (GES) 2
1.2 L'oxyde nitreux 4 1.3 L'échantillonnage à flux passif 5
1.4 L'échantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005) 8
1.4.1 Description de l'échantillonneur 8 1.4.2 Procédure de mesure d'émissions 11
1.4.3 Principes théoriques 13 1.4.4 Identification des problèmes de l'échantillonneur 16
2. Hypothèses, but et objectifs 18
3. Méthodologie 19 3.1 Volet 1 : Amélioration de la performance de l'échantillonneur 19
3.1.1 Étanchéisation de l'échantillonneur 19
3.1.2 Retrait du diffuseur 22 3.1.3 Réduction de l'excès de N2O 23
3.1.4 Installation de la plaque à orifice 24
3.1.5 Mode opératoire 24 3.1.5.1 Conditionnement de l'adsorbant 24
3.1.5.2 Protocole de l'expérience d'adsorption 26 3.1.5.3 Protocole de l'expérience de désorption 29
3.1.6 Dispositif expérimental 31 3.1.7 Exactitude et précision de l'échantillonneur 32
3.2 Volet 2 : Augmentation de la capacité d'adsorption 33 3.2.1 Augmentation de la capacité de stockage de la zéolite dans
l'échantillonneur 33 3.2.2 Mode opératoire 35 3.2.3 Plan expérimental 35 3.2.4 Exactitude et précision de l'échantillonneur 36
3.3 Volet 3 : Étude d'une nouvelle procédure de désorption 36
3.3.1 Procédure de désorption étudiée 36
3.3.2 Mode opératoire 37 3.3.3 Dispositif expérimental 37 3.3.4 Évaluation des procédures de désorption 38
3.4 Volet 4 : Détermination des constantes de calibration (K) 38
3.4.1 Mode opératoire 38 3.4.2 Dispositif expérimental 40
4. Résultats et discussion 41 4.1 Volet 1 : Performance de l'échantillonneur modifié 41
4.1.1 Résultats de l'expérience 41 4.1.2 Efficacité d'adsorption 42 4.1.3 Efficacité de désorption 43 4.1.4 Exactitude et précision 45 4.2 Volet 2 : Augmentation de la capacité d'adsorption 46
4.2.1 Résultats de l'expérience 46 4.2.2 Efficacité d'adsorption 48 4.2.3 Efficacité de désorption 49 4.2.4 Exactitude et précision 51 4.3 Volet 3 : Étude d'une nouvelle procédure de désorption 52
4.3.1 Adsorption 52 4.3.2 Désorption 54 4.3.3 Exactitude et précision 57
4.4 Volet 4 : Calibration des échantillonneurs 59
Conclusions 61 Recommandations 65 Bibliographie 67 ANNEXE A. Schéma de l'échantillonneur et de ses composants 72
ANNEXE B. Résultats statistiques 74 ANNEXE C. Données de l'expérience de calibration des échantillonneurs 86
Liste des tableaux
Tableau 1. Propriétés de l'oxyde nitreux 4 Tableau 2. Plan expérimental pour l'étude de la performance de l'échantillonneur 31
Tableau 3. Plan expérimental pour l'étude de l'influence de l'augmentation de la
quantité de zéolite sur la performance de l'échantillonneur 35 Tableau 4. Plan expérimental pour l'étude de la procédure de désorption proposée 37
Tableau 5. Plan expérimental pour la détermination des constantes de calibration 40
Tableau 6. Résultats de la performance de l'échantillonneur modifié 41 Tableau 7. Tableau des résultats des tests de F sur les effets fixes 43
Tableau 8. Tableau ANOVA des résultats du test de F sur l'effet fixe « N20 total
injecté » 44 Tableau 9. Temps de saturation des couches de zéolite pour trois concentrations de
N20 46
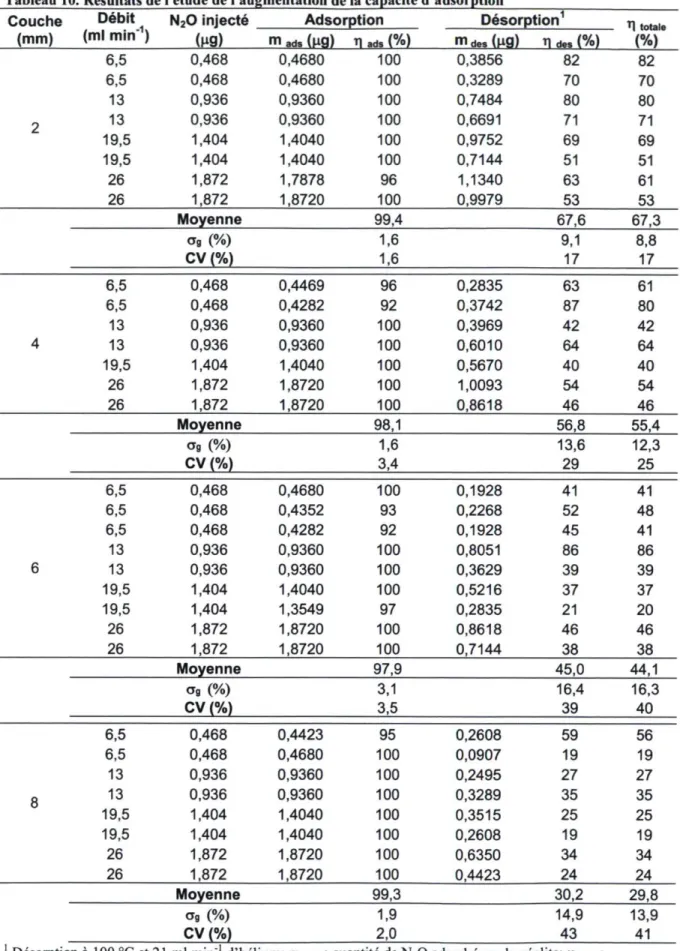
Tableau 10. Résultats de l'étude de l'augmentation de la capacité d'adsorption 47 Tableau 11. Tableau ANOVA des résultats du test de F sur les effets fixes 49
Tableau 12. Tableau des tests de F sur les effets fixes 50 Tableau 13. Performance de l'échantillonneur modifié avec la nouvelle procédure
de désorption 53 Tableau 14. Tableau des tests de F de l'essai de désorption avec la procédure #2 54
Tableau 15. Tableau des tests de F de l'essai de deux procédures de désorption 57 Tableau 16. Comparaison de l'exactitude et de la précision pour les deux procédures
de désorption 57 Tableau 17. Constantes de calibration des échantillonneurs 59
Tableau BI. Tableaux des résultats des tests de F 75 Tableau B2. Tableaux des résultats des tests de F 76 Tableau B3. Tableau des contrastes entre les quantités de N2O injectées 77
Tableau B4. Tableaux des résultats des tests de F 78 Tableau B5. Estimateurs des composantes de variance associées aux effets
aléatoires 80 Tableau B6. Tableau des résultats des tests de F sur les effets fixes 80
Tableau B7. Tableau de contrastes entre les niveaux 81 Tableau B8. Tableau des résultats des tests de F sur les effets fixes de la procédure
de désorption #2 83 Tableau B9. Contrastes entre les niveaux 83
Tableau B10. Calcul de dj/a pour élimination de données selon les critères de
Chauvenet 84 Tableau BI 1. Critères de Chauvenet pour le rejet de données 84
Tableau Cl. Vitesses de l'air calibrées dans le tunnel de vent 87 Tableau C2. Vitesse de l'air dans les échantillonneurs avec une couche d'adsorbant
de 2mm d'épaisseur 87 Tableau C3. Vitesse de l'air dans les échantillonneurs avec une couche d'adsorbant
Tableau C4. Vitesse de l'air dans les échantillonneurs avec une couche d'adsorbant
de 6mm d'épaisseur 91 Tableau C5. Vitesse de l'air dans les échantillonneurs avec une couche d'adsorbant
Liste des figures
Figure 1. Processus de diffusion 6 Figure 2. Échantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005) 9
Figure 3. Pièces principales de l'échantillonneur de Godbout et al. (2006) et
Gaudet (2005) 9 Figure 4. Schéma des pièces de la cartouche (Gaudet, 2005) 10
Figure 5. Échantillonneur, orifice et connecteur 11 Figure 6. Facteur K de l'échantillonneur avec des plaques à orifice de différentes
dimensions; adaptée de Gaudet (2005) 12 Figure 7. Techniques d'étanchéisation, A : application d'un scellant de filets de
conduite, B : installation de rondelles plates en cuivre et C : installation
de rondelles en Viton 20 Figure 8. Vecteurs de vitesse de l'air dans l'échantillonneur colorés par magnitude
(m s"1) et axisymétriques à l'axe X 22
Figure 9. Plaque à orifice accouplée au raccord 24 Figure 10. Système de conditionnement des échantillonneurs 25
Figure 11. Système d'adsorption directe de N2O 26
Figure 12. Système de désorption 29 Figure 13. Schéma des nouveaux grillages métalliques 34
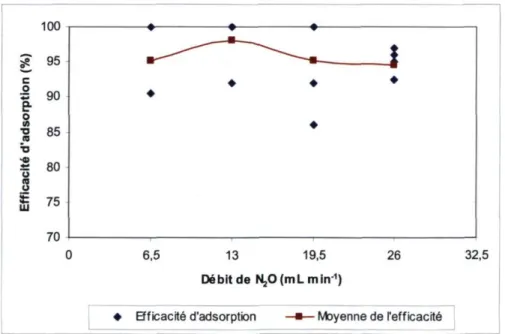
Figure 14. Tunnel de vent pour calibrer les échantillonneurs 38 Figure 15. Efficacité d'adsorption de l'échantillonneur modifié en fonction du
débit 42 Figure 16. Moyennes et limites de confiance à 90 % pour l'efficacité de
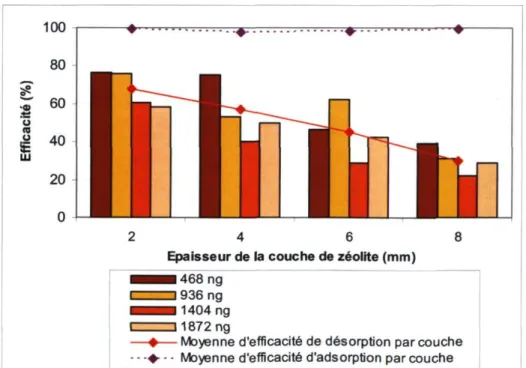
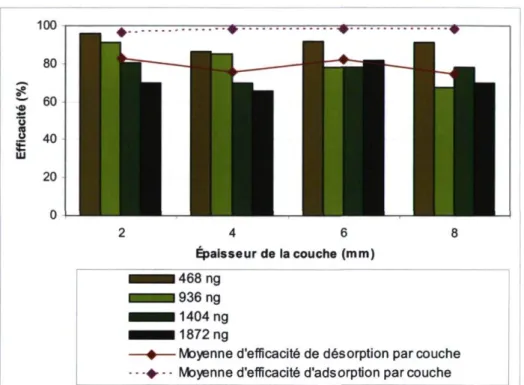
désorption, par N2O injecté 44 Figure 17. Efficacité d'adsorption par couche de zéolite 48
Figure 18. Efficacité de désorption moyenne par quantité de N2O dirigé vers
l'échantillonneur 49 Figure 19. Efficacités de désorption moyenne de la nouvelle procédure de
désorption (procédure #2) 54 Figure 20. Comparaison de l'efficacité de désorption moyenne des procédures de
désorption 1 et 2 56 Figure A 1. Schéma de l'échantillonneur de Godbout et al. (2006) et Gaudet
(2005) modifié 73 Figure B1. Moyennes et limites de confiance à 90 % pour l'efficacité d'adsorption
par débit 76 Figure B2. Moyennes et limites de confiance à 90 % pour l'efficacité d'désorption
par débit 77 Figure B3. Moyennes et limites de confiance à 90 % pour l'efficacité d'adsorption
par débit 79 Figure B4. Moyennes et limites de confiance à 90 % pour l'efficacité d'adsorption
par épaisseur de couche de zéolite 79 Figure B5. Graphique de résidus en fonction du débit 80
Figure B6. Moyennes et limites de confiance à 90 % pour l'efficacité de
désorption par quantité de N2O injectée 82 Figure B7. Moyennes et limites de confiance à 90 % pour l'efficacité de
désorption par épaisseur de couche de zéolite 82 Figure B8. Moyennes et limites de confiance à 95 % pour l'efficacité de
désorption par quantité de N2O injectée 85 Figure B9. Graphique de résidus en fonction du temps 85 Figure Cl. Calibration de l'échantillonneur C avec une couche d'adsorbant de
2mm d'épaisseur 87 Figure C2. Calibration de l'échantillonneur D avec une couche d'adsorbant de
2mm d'épaisseur 88 Figure C3. Calibration de l'échantillonneur E avec une couche d'adsorbant de
2mm d'épaisseur 88 Figure C4. Calibration de l'échantillonneur Y avec une couche d'adsorbant de
2mm d'épaisseur 88 Figure C5. Calibration de l'échantillonneur C avec une couche d'adsorbant de
4mm d'épaisseur 89 Figure C6. Calibration de l'échantillonneur D avec une couche d'adsorbant de
4mm d'épaisseur 90 Figure C7. Calibration de l'échantillonneur E avec une couche d'adsorbant de
4mm d'épaisseur 90 Figure C8. Calibration de l'échantillonneur Y avec une couche d'adsorbant de
4mm d'épaisseur 90 Figure C9. Calibration de l'échantillonneur C avec une couche d'adsorbant de
6mm d'épaisseur 91 Figure C10. Calibration de l'échantillonneur D avec une couche d'adsorbant de
6mm d'épaisseur 92 Figure C i l . Calibration de l'échantillonneur E avec une couche d'adsorbant de
6mm d'épaisseur 92 Figure Cl2. Calibration de l'échantillonneur Y avec une couche d'adsorbant de
6mm d'épaisseur 92 Figure Cl3. Calibration de l'échantillonneur C avec une couche d'adsorbant de
8mm d'épaisseur 93 Figure Cl4. Calibration de l'échantillonneur D avec une couche d'adsorbant de
8mm d'épaisseur 94 Figure Cl5. Calibration de l'échantillonneur E avec une couche d'adsorbant de
8mm d'épaisseur 94 Figure Cl6. Calibration de l'échantillonneur Y avec une couche d'adsorbant de
Introduction
Actuellement, les gaz à effet de serre (GES) sont devenus le centre d'attention du monde entier à cause de leurs conséquences sur le réchauffement global. Les politiques mises en place pour réduire les émissions de certains pays ont été efficaces jusqu'à un certain point, mais pas suffisamment pour faire le contrepoids à la croissance mondiale des émissions (GreenFacts, 2007). L'agriculture est un des secteurs les plus importants en tant qu'émetteur d'oxyde nitreux (N2O). Ce secteur a cependant un grand potentiel pour réduire les émissions de ce gaz dans l'atmosphère.
Il est nécessaire de développer des méthodes rapides, efficaces et économiques pour mesurer les gaz émis par l'agriculture. Pour des sources à faibles émissions, les approches conventionnelles sont souvent trop sophistiquées et coûteuses. L'échantillonnage à flux passif a démontré depuis plusieurs années qu'il est un outil approprié pour mesurer les émissions de faible intensité.
La présente recherche a pour but d'optimiser l'échantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005) et d'améliorer les méthodes d'analyse afin de mesurer de faibles émissions de N2O. L'étude vise aussi à déterminer l'erreur de mesure et, finalement, à proposer des recommandations pour de futurs travaux de recherche.
1. Mise en contexte
1.1 Émission de gaz à effet de serre (GES)
Une partie de l'énergie apportée par le soleil à la terre est transformée en chaleur et émise vers le ciel sous forme infrarouge. Les gaz à effet de serre (GES) présents dans l'atmosphère absorbent le rayonnement infrarouge émis par la surface de la terre et le redirige dans toutes les directions, y compris vers la terre. Par conséquent, la température de la planète augmente en produisant un réchauffement global. Ce phénomène est connu comme l'effet de serre.
La principale raison de la hausse des températures est liée à l'augmentation des GES dans l'atmosphère. Le siècle et demi d'industrialisation a contribué au dégagement des GES par la combustion de quantités de plus en plus grandes de pétrole, d'essence et de charbon, par la coupe des forêts ainsi que par certaines pratiques agricoles (CCNUCC, 2008).
Les émissions mondiales de gaz à effet de serre ont augmenté considérablement depuis l'époque préindustrielle. Entre 1970 et 2004, elles ont augmenté de 70 %, soit de 28,7 à 49 Gigatonnes équivalents de dioxyde de carbone (Gt éq. CO2) (IPCC, 2007).
Le Protocole de Kyoto de la CCNUCC (2008) est une entente créée en 1997 où 38 pays industrialisés, dont le Canada, se sont engagés à réduire leurs émissions de GES. Cet accord, entré en vigueur en février 2005, a pour objectif de stabiliser la concentration des GES, et conséquemment, de promouvoir le développement durable. Les six principaux GES visés par le protocole sont : le dioxyde de carbone (CO2), le méthane (CH4), l'oxyde nitreux (N20) les hydrofluorocarbones (HFC), les hydrocarbures perfluorés (PFC) et
l'hexafluorure de soufre (SFÔ).
La croissance des émissions des GES fait passer la température globale de la planète à des sommets si élevés qu'elle crée des désordres climatiques et engendre de grands changements climatiques. Les conséquences qui pourraient découler d'une augmentation importante des concentrations des GES prennent parfois l'allure d'un scénario catastrophique, dont quelques impacts ont déjà été observés. Le Groupe d'experts
intergouvememental sur l'évolution du climat (IPCC, 2007) a déclaré que le doublement des concentrations se traduira par une plus grande vulnérabilité de la santé humaine, des écosystèmes et de divers secteurs de l'activité économique.
Les GES provoquent des impacts d'intensité différente sur le réchauffement de la planète. Par conséquent, leurs émissions sont habituellement exprimées en équivalents de CO2 (éq. CO2) pour permettre de comparer leurs effets relatifs. Par exemple, 1 kg de N2O produit le même réchauffement qu'environ 310 kg de CO2 (sur une période de 100 ans). C'est pour cette raison que le N2O vaut donc 310 équivalents de CO2; de même, 1 kg de CH4 représente 21 équivalents de CO2 (Janzen et al., 1999).
En ratifiant le Protocole de Kyoto, le Canada s'est engagé à réduire ses émissions de GES de 6 % par rapport au niveau de 1990, d'ici 2012. Environnement Canada (2008), dans son inventaire canadien des gaz à effet de serre, signale que les émissions totales de gaz à effet de serre au Canada pour 2006 ont été de 721 Mt éq. CO2, ce qui représente une augmentation de 22 % par rapport aux 592 Mt éq. CO2 émises en 1990.
Le secteur de l'énergie, de l'industrie, de l'agriculture, des déchets et des solvants sont les principales sources d'émission de GES établies par le protocole de Kyoto. Agriculture et Agroalimentaire Canada (AAC, 2006) signale qu'en 2004, le secteur de l'agriculture était responsable d'environ 7,2 % des émissions de GES au Canada, dont les plus importants sont le N20, le CH4 et le C02.
En 2006, le gouvernement du Québec a rendu public son plan d'action pour atteindre les objectifs de Kyoto. Le plan québécois prévoit une réduction de 1,5 % des émissions de GES par rapport aux niveaux de 1990 (Greenpeace Canada, 2006), soit 86,4 Mt éq. CO2 (Houle et al., 2002). Les émissions totales de GES du Québec en 2003 se chiffraient à 91,0 Mt éq. CO2 et représentaient 12,3 % des émissions canadiennes. La même année, le secteur agricole au Québec a contribué à 9,4 % des émissions de GES, soit 8,5 Mt éq. CO2 (MDDEP, 2006a). Pour le secteur agricole, le GES le plus émis est le N2O. En effet, 50 à 80 % des émissions totales canadiennes sont du N2O, suivi par le CH4 (30 %) et le C02 (<1 %) (Nature Québec / UQCN, 2006).
1.2 L'oxyde nitreux
L'oxyde nitreux (N2O) est aussi connu sous le nom de protoxyde d'azote ou gaz hilarant. Il est constitué de deux atomes d'azote et un atome d'oxygène. Ce gaz appartient au groupe des oxydes d'azote (NxOy). Ce groupe fait référence à plusieurs composés
chimiques binaires gazeux formés par la combinaison de l'oxygène et l'azote pouvant être très polluants.
L'oxyde nitreux est naturellement présent dans l'atmosphère en très faible quantité (environ 0,3 ppmv), mais cette concentration s'accroît d'environ 0,3 % par an. Une grande partie de cette augmentation est causée par l'agriculture, qui produit jusqu'à 70 % du total des émissions de N2O d'origine anthropique (Janzen et al., 1999).
Dans la haute atmosphère, l'oxyde nitreux est converti en oxyde nitrique (NO). Celui-ci décompose l'ozone (O3), qui a comme principal rôle de filtrer les rayons ultraviolets émis par le soleil. Par conséquent, le N20 est considéré comme un des GES les plus importants
et puissants. Le potentiel de réchauffement de ce gaz est 310 fois supérieur à celui du CO2 et sa durée de vie dans l'atmosphère est d'environ 120 ans. Les principales propriétés du N2O sont présentées au tableau 1.
Tableau 1. Propriétés de l'oxyde nitreux
Propriété Valeur Unité Source
Poids moléculaire
Densité (1,013 bar et 21 °C)
Gravité spécifique (air = 1) (1,013 bar et 21 °C) Volume spécifique (1,013 bar et 21 °C)
Viscosité (1,013 bar et 0 °C)
Conductivité thermique (1,013 bar et 0 °C) Solubilité dans l'eau (1,013 bar et 5 °C) Diamètre moléculaire
Durée de vie atmosphérique
Facteur de réchauffement global (CQ2=1)
44,013 g/mol 1 1,836 kg/m3 2 1,53 0,543 m3/kg 0,000136 Poise 14,57 mW/(m.K) 1,14 vol/vol 0,33 nm 3 120 année 4, 5 et 6 310 7
Sources : 1. AIR LIQUIDE © (2009) 2. http://www.airproductS.com/ Air Products and Chemicals, Inc. (2003) 3. Breck (1974) cité par Godbout et al. (2006) 4. Houghton, et al (1997) 5. Abrous et Hbiak (2005) 6. Nature Québec / UQCN (2006) 7. RecyConsult © (2000)
Au Québec, entre 1990 et 2003, les émissions de N2O ont augmenté de 13,6 %, passant de 3,12 Mt éq.C02 à 3,55 Mt éq.C02 (MDDEP, 2006a), où le secteur agricole a été
l'émetteur le plus important de N2O. Agriculture et Agro-alimentaire Canada (Janzen et al., 1999) classifie les émissions de N2O de l'industrie agricole en trois catégories :
1. Émissions directes par le fumier animal : La quantité de gaz émise dépend de la méthode de gestion, des propriétés du fumier, des espèces animales et du nombre d'animaux. Les émissions québécoises produites par cette activité agricole ont augmenté de 10,4 % entre 1990 et 2003, passant de 2,45 Mt éq. C02 à 2,70 Mt éq.
C02 (MDDEP, 2006b).
2. Émissions directes par les sols agricoles : Les émissions directes de N2O des sols proviennent des engrais, des fumiers minéraux et organiques, des légumineuses et des résidus de cultures (Janzen et al., 1999).
3. Émissions indirectes : Les émissions indirectes de N2O sont généralement produites à partir de l'azote émis par les systèmes agricoles. Cet azote peut se dissiper par voie aérienne (volatilisation d'ammoniac et émissions de NOx) ou par voie souterraine (lessivage de nitrates) et être redéposé sous forme sèche ou humide (Agu et al., 2000). Au Québec, les émissions indirectes de N2O ont été estimées à 4 Gg en 1996, ce qui équivaut à 1,18 Mt éq. CO2. Cette valeur montre que les émissions indirectes sont aussi importantes que les émissions directes.
1.3 L'échantillonnage à flux passif
L'échantillonnage à flux passif est une technique permettant la mesure de l'émission ou de la concentration d'un gaz ciblé. Cette technique repose sur la diffusion du gaz à travers une surface diffusive constitué d'un adsorbant (Sigma-Aldrich Co, 2008) ou sur l'infiltration du gaz à travers une membrane semi-perméable ou un milieu poreux (Kot-Wasik et al, 2007). L'adsorbant retient les gaz ciblés et les libère au moyen d'un procédé de désorption thermique ou chimique.
Les échantillonneurs à flux passif n'ont pas besoin d'instruments pour contrôler le taux d'extraction des gaz durant l'échantillonnage. La nature passive de cette technique permet que le taux d'extraction soit contrôlé d'une façon physique, soit par une couche d'air statique (Kumagai et Koda, 1999), soit par l'infiltration à travers une membrane (Namiessnik et al., 2005 et Gorecki et Namiesnik 2002), ou soit par la diffusion gazeuse à travers un matériel poreux adsorbant (Sigma-Aldrich Co., 2008), ainsi que la combinaison des deux dernières (Figure 1).
. . a *
Surface diffusive
Surface absorbante
Figure 1. Processus de diffusion Adaptée de Sigma-Aldrich Co. (2008)
Les échantillonneurs à flux passif déterminent l'émission moyenne de substances gazeuses par rapport au temps d'adsorption. Cela signifie que le gaz collecté est accumulé dans l'adsorbant pendant tout le prélèvement. Ainsi, la méthode d'échantillonnage passive ne subit pas les variations des concentrations extrêmes ou accidentelles (Namiessnik et al., 2005). Cette caractéristique est référencée dans la littérature comme « Time-Weighted Average ou TWA » (Kot-Wasik et al., 2007, Huckins et al., 2006, Namiesnik et al., 2005, Ni et Heber, 2001, Kumagai et Koda, 1999 et May, 1989).
En comparaison avec les autres techniques de mesure d'émissions, l'utilisation d'échantillonneurs à flux passif présente les avantages suivants:
- leurs principes de fonctionnement sont fondés sur une technique robuste et simple;
- les résultats ne sont pas affectés par les variations de concentration ou les concentrations extrêmes du gaz ciblé lors de l'échantillonnage,
- ils utilisent des matériaux inertes qui assurent l'absence de réactions entre l'adsorbant et les échantillons, ce qui permet à ces derniers de rester inaltérés dans l'échantillonneur durant plusieurs mois après l'échantillonnage;
- ils réduisent les frais reliés aux analyses et fournissent un rapport coût-bénéfice intéressant;
- ils n'utilisent pas de système de pompage pour l'adsorption;
- puisque ils ne requièrent aucune source d'énergie lors de l'échantillonnage, ils deviennent très avantageux sur des sites où l'électricité n'est pas disponible; - la préparation, le conditionnement et l'installation requis sont faciles; - ils ne nécessitent pas de surveillance intensive;
- ils sont légers, silencieux et discrets; - leurs composants sont réutilisables;
- ils peuvent être manipulés et transportés facilement; - ils sont constitués de matériaux ininflammables.
Toutefois, il existe des facteurs qui affectent la performance de l'échantillonnage passif, tels que:
- Le temps d'échantillonnage:
Le temps limite d'échantillonnage ou d'exposition est déterminé à partir du temps que dispose l'absorbant avant d'atteindre la saturation. Le temps de saturation est influencé aussi bien par les propriétés de l'absorbant que par les conditions de l'environnement de l'échantillonnage. Si le temps limite d'exposition est dépassé, l'adsorbant est saturé et le résultat obtenu sous-estimera l'émission réelle. Cette situation peut être évitée en calculant, avec un facteur de sécurité suffisamment rigoureux le temps d'échantillonnage.
- La capacité de sélection des gaz ciblés:
Lors de l'échantillonnage, les adsorbants sont susceptibles d'adsorber des substances autres que les gaz désirés. La concentration de ces substances dans l'adsorbant réduit l'efficacité et le temps d'adsorption de l'échantillonneur.
- Les conditions environnementales :
Plusieurs auteurs tels que MARKES International (2008), Kim et al. (2007), Kot-Wasik et al., (2007), Yu (2006), Namiessnik et al., (2005), Varshney et Singh (2003), Harper (1993 et 2000), Kumagai et Koda (1999), Brancaleonia et al. (1999), 3M (1996) et Fiorito et Harper (1995) ont étudié l'effet de différents paramètres environnementaux sur la capacité d'adsorption des échantillonneurs à flux passif, spécialement l'humidité et la température du milieu d'échantillonnage. Aussi, quelques auteurs (Welch, 2003, Gaudet, 2005, Kumagai et Koda, 1999, Varshney et Singh, 2003, Thomas et al., 2006) ont évalué l'influence de la vitesse et de l'angle d'entrée de l'air sur l'efficacité d'adsorption des échantillonneurs à flux passif. Parfois, ces facteurs peuvent diminuer considérablement la capacité et le temps d'adsorption de l'échantillonneur. Donc, il est important d'étudier l'effet de ces éléments sur la performance des échantillonneurs.
1.4 L'échantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005) 1.4.1 Description de l'échantillonneur
Godbout et al. (2006) et Gaudet (2005) ont développé un échantillonneur à flux passif et un procédé destinés à la mesure des émissions du N2O et du CH4 provenant de sources
agricoles. Cet échantillonneur peut être décrit comme suit : - Environnement d'échantillonnage : milieux gazeux; - Mode de lecture : indirecte;
- Géométrie de l'échantillonneur : type tube avec adsorption axiale;
- Principe d'extraction : adsorption à partir d'un matériel solide semi-perméable; - Type de désorption : thermique.
Puisqu'il n'existe pas de classification standard, les critères de classification déterminés ci-haut ont été identifiés dans la littérature à partir des principales caractéristiques des échantillonneurs à flux passif.
L'échantillonneur de Godbout et al. (2006) et Gaudet (2005) a une longueur de 175 mm et un diamètre de 50 mm (figure 2). Il est en acier inoxydable et est constitué de trois pièces principales (figure 3). Le cône antérieur (figure 3, pièce 1) est constitué d'un tube de 6,35 mm (% de po.) de diamètre correspondant à l'entrée d'air suivie d'une section conique avec une ouverture graduelle de 14,6°. Lorsque l'air sort du tube, une partie de ce fluide a tendance à suivre le contour intérieur du cône. La mécanique des fluides nomme ce phénomène « effet Coanda ». Cela permet au gaz de se répandre dans le cône pour traverser une plus grande surface de l'adsorbant.
Figure 2. Echantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005)
Figure 3. Pièces principales de l'échantillonneur de Godbout et al. (2006) et Gaudet (2005)
La cartouche (figure 3, pièce 2) est la pièce centrale de l'échantillonneur qui unit les cônes antérieur et postérieur. Elle contient l'adsorbant. La figure 4 présente les pièces
composant la cartouche. Deux grillages métalliques (figure 4, pièces cl et c2) et deux anneaux de soutien (figure 4, pièces a et e) retiennent l'adsorbant. Un diffuseur conique placé au centre de l'aire d'adsorption (figure 4, pièce b) contraint l'air à occuper un espace maximal dans la cartouche et vise à améliorer l'uniformité de la vitesse de l'air (Gaudet 2005). Il est retenu grâce à deux vis dans sa partie postérieure (Figure 4, pièces dletd2).
(c2) <d2) (e)
Cartouche assamblée
Figure 4. Schéma des pièces de la cartouche (Gaudet, 2005)
Le matériel utilisé comme adsorbant dans l'échantillonneur développé par Godbout et al. (2006) et Gaudet (2005) est le tamis moléculaire de zéolite 5A. La zéolite est un matériel minéral caractérisé par sa structure poreuse capable de retenir des molécules ayant une grandeur plus petite que le diamètre de ses pores. La dimension des pores de la zéolite 5A est de 5 Angstrom (5xl0~7 mm). Comme la molécule de N2O a 3,3 Angstrom de diamètre
1. Le cône postérieur (figure 3, pièce 3) est la dernière pièce de l'échantillonneur. Il a pour fonction de diriger l'air vers l'extérieur au travers un tuyau de 6,35 mm (% po.) de diamètre.
Dans le but de contrôler et de réduire le débit d'air dans l'échantillonneur, une petite plaque circulaire avec un orifice de 0,52 mm est installée à la sortie de l'échantillonneur, juxtaposée au cône postérieur (figure 5). Cet orifice réduit le flux de gaz et permet non seulement d'avoir une linéarité dans la relation entre la vitesse de l'air extérieur et intérieur, mais aussi d'échantillonner des sources d'émission de concentrations plus élevées ou d'augmenter le temps d'échantillonnage. La figure 6 présente la relation entre la vitesse d'air extérieur et celle de l'intérieur pour l'échantillonneur sans plaque à orifice et avec une plaque à orifice de 1 mm et de 0,52 mm de diamètre.
Figure 5. Échantillonneur, orifice et connecteur
1.4.2 Procédure de mesure d'émissions 1.4.2.1 Calibration de l'échantillonneur
La calibration de l'échantillonneur consiste à déterminer la relation entre la vitesse de l'air qui passe à travers l'échantillonneur et la vitesse de l'air qui circule autour. Gaudet (2005) a déterminé que cette relation varie entre 0,0036 et 0,0039 avec des coefficients de corrélation (K) entre 0,9923 et 0,9774 pour quatre échantillonneurs. Ce facteur est utilisé pour le calcule de l'émission suite à l'échantillonnage.
2 3 4 s e 7 e Vitesse de l'air autour de l'échantillonneur (m/s)
Sans orifice Orifice de 1 mm Orifice de 0,52 mm
— Tendance (sans orifice) — Tendance (1 mm)
- Tendance (0,52 mm)
Figure 6. Facteur K de l'échantillonneur avec des plaques à orifice de différentes dimensions; adaptée de Gaudet (2005)
1.4.2.2 Conditionnement
Le but du conditionnement est de purger la zéolite et de préparer les échantillonneurs pour le prélèvement. La zéolite requière l'action de la chaleur et un débit de gaz inerte qui la traverse pour extraire les molécules de gaz présentes dans ses pores. MARKES international (2002) recommande de chauffer la zéolite à 350 °C pendant 2h ou à 300 °C pendant une nuit tout en appliquant un débit d'au moins 50 ml min"1 d'azote ou d'hélium.
Gaudet (2005) a appliqué un débit de 2,4 L min"1 d'azote à quatre échantillonneurs
chauffés à 350 °C pendant 150 min. 1.4.2.3 Adsorption ou échantillonnage
Cette étape consiste à exposer l'échantillonneur à une source de gaz ciblés durant un temps spécifique afin d'en déterminer l'émission. Il est important que l'échantillonneur soit étanche aussi bien avant qu'après le prélèvement car, dans le cas contraire, la zéolite
continue son processus d'adsorption et les résultats sont ainsi altérés. Gaudet (2005) a conclu que la zéolite contenue dans l'échantillonneur est capable d'adsorber 91 % (± 11 %) du N2O qui passe au travers. Cependant, il a aussi montré que la zéolite n'adsorbe pas facilement le CH4. Seulement 9 % (± 6 %) du CH4 qui traverse l'échantillonneur est adsorbé.
1.4.2.4 Désorption et analyse
Ces dernières étapes visent à extraire le gaz récolté pendant le prélèvement au moyen d'une désorption thermique en laboratoire. Cette technique utilise la chaleur pour augmenter la volatilité des gaz d'une matrice solide et le flux d'un gaz inerte qui passe à travers la matrice pour entraîner les gaz volatilisés. Les gaz sortant de l'échantillonneur sont récoltés de telle sorte qu'ils puissent être quantifiés. La chromatographie en phase gazeuse est la méthode couramment utilisée pour la détermination de la concentration des gaz échantillonnés.
Puisque le processus est contrôlé par un transfert de masse, des facteurs comme la température, le temps de chauffage et la quantité de gaz inerte sont des éléments clés dans l'optimisation de l'efficacité de désorption. Godbout et al. (2006a) recommandent une température de 100 °C pour réaliser la désorption du N20 de la zéolite. Gaudet (2005) a
employé un flux de 21 ml min"1 d'azote pendant 40 min et une température de chauffage
de 100 °C. MARKES International (2002) recommande 165 °C et un flux de 25 ml min"1
pendant un temps de 3 à 5 minutes. Cependant, même si l'azote a été généralement utilisé comme gaz transporteur (Gaudet, 2005; Godbout et al. 2006 and 2006b; Kumagai & Koda, 1999), l'hélium a aussi été utilisé avec ces derniers paramètres (Henderson et al., 2003; MARKES International, 2002 et Park, et al., 1998).
1.4.3 Principes théoriques
L'échantillonneur de Godbout et al. (2006) et Gaudet (2005) est basé sur le concept original de l'échantillonneur de Ferm (1986) et les principes théoriques de Scholtens et al. (2003) et Schjoerring et al. (1992).
Le fonctionnement de l'échantillonneur passif est fondé sur deux principes. Premièrement, la circulation de l'air autour d'un corps provoque une réduction de pression en aval de l'échantillonneur. Cette perte de pression provoque l'écoulement dans l'échantillonneur. Elle peut être décrite par l'équation suivante :
A PD= ^ f = \ cDp v2 m [1]
Ap 2
Où : L\PD = Différence de pression causée par le corps de l'échantillonneur, Pa
FD = Force de résistance à la surface de l'échantillonneur, N
CD = Coefficient de friction, adimensionnel
p = Densité de l'air, kg m"3
vm = Vitesse de l'air après l'échantillonneur, m s"1
Ap = Aire du corps projetée perpendiculairement au flux de l'air, m2
Deuxièmement, par l'effet que la circulation de l'air à travers une plaque à orifice dépend de la perte de pression causée par la restriction du flux :
A P° = Y ~ CT2P V 2°=\C°P V 2° [2]
Où : AP0 = Perte de pression causée par l'orifice, Pa
B = Relation entre le diamètre de l'orifice et celui du conduit, adimensionnelle Y = Facteur d'expansion, adimensionnel
C = Coefficient de débit, adimensionnel C0 = Constante de l'orifice, adimensionnelle
v0= Vitesse de l'air à travers l'orifice, m s"1
La perte de pression dans les équations [1] et [2] dépend du carré de la vitesse de l'air. Donc, sous des conditions atmosphériques normales, ces deux équations peuvent être égalées (PD = P0). À cet égard, la combinaison des équations [1] et [2] peut être
v
°
=y ~ c
L Vm = K°
Vm [3]Où Ks est la constant adimensionnelle de calibration de l'échantillonneur.
Puisque les constants Co et Co sont fonction de la vitesse de l'air à l'intérieur et à l'extérieur de l'échantillonneur, l'équation [3] peut être réécrite comme suit (Gaudet, 2005):
Ks= f = [4]
vm Vcosa
Où a est l'angle formé par l'axe parallèle au flux et l'axe de la direction du vent.
La constante de calibration peut également être déterminée à partir de la vitesse de l'air dans le tuyau de sortie de l'échantillonneur en remplacement de la vitesse de l'air dans l'orifice. Dans ce cas, l'équation 4 est réécrite ainsi :
K = f = [5] vm Vcosa
Où : K = Constante de calibration de l'échantillonneur à partir de la vitesse de l'air dans le tuyau de sortie, adimensionnelle
vs = Vitesse de l'air dans le tuyau de sortie de l'échantillonneur, m s"1
Les équations [4] et [5] montrent théoriquement la relation linéaire entre les vitesses de l'air à l'intérieur et à l'extérieur de l'échantillonneur. Dans le but de valider l'utilisation de l'échantillonneur passif dans des conditions réelles, ces relations doivent rester constantes pour toute variation de la vitesse de l'air. Ainsi, la nécessité de mesurer la vitesse de l'air sur le terrain sera éliminée.
Finalement, après l'échantillonnage et la mesure du gaz adsorbé, l'émission est déterminée au moyen de l'équation suivante :
M M
F = ; = ; [6] n r2K , M n R2 K At
Où : F = Flux du gaz, g m"2 s"1
M = Masse de gaz ciblé collecté, g r = Rayon de l'orifice, m
K. = Constante de calibration de l'échantillonneur à partir de la vitesse de l'air dans l'orifice, adimensionnelle
R = Rayon du tuyau de sortie, m
K = Constante de calibration de l'échantillonneur à partir de la vitesse de l'air dans le tuyau de sortie, adimensionnelle
At = Temps d'échantillonnage, s
1.4.4 Identification des problèmes de l'échantillonneur 1.4.4.1 Efficacité à la désorption :
Une grande partie de l'erreur systématique déterminée par Gaudet (2005) est reliée à la procédure de désorption. Les échantillonneurs sont connectés à une série de tuyaux qui sont remplis d'air avant de commencer la désorption. La zéolite adsorbe le N2O de cet air et contamine conséquemment les échantillons, entrainant une surestimation de 134 %. Gaudet (2005) propose un ajustement aux données mesurées, à savoir de soustraire de la masse de gaz de N2O mesuré, la quantité de N2O que la zéolite a pu adsorber par contamination. Cependant, les résultats corrigés continuent d'être plus grands que 100 %, soit 108 %.
1.4.4.2 Temps d'échantillonnage
L'échantillonneur à flux passif de Godbout et al. (2006) et Gaudet (2005) utilise une méthode d'échantillonnage de type cumulatif. Donc, l'émission moyenne d'une source de gaz est déterminée à partir du ratio entre la quantité adsorbée et le temps d'exposition de l'échantillonneur. Ainsi, la mesure d'une émission de gaz est plus représentative avec des temps d'échantillonnage longs sans que soit atteinte la saturation du matériel adsorbant (par exemple : 6 heures, 12 heures, 1 journée, etc.). Selon Godbout et al. (2006), le temps d'échantillonnage est lié directement à la capacité d'adsorption. Si cette capacité est augmentée, l'échantillonneur peut fonctionner avec des temps d'échantillonnage plus
longs avant d'atteindre la saturation. Gaudet (2005) signale que lorsque l'échantillonneur est exposé à une source de CH4 de 10 ppm à 15 ml min"1, la zéolite placée à l'intérieur de
l'échantillonneur (700 mg) atteint la saturation en 56 minutes. D'autre part, si l'échantillonneur est exposé à une source de N2O de 2 ppm à 15 ml min"1, la saturation de
la zéolite sera atteinte en 50 minutes. 1.4.4.3 Étanchéité
Des fuites dans les échantillonneurs ont été détectées. L'étanchéité de l'échantillonneur est un paramètre important afin d'assurer l'exactitude et la précision des mesures. La non-étanchéité amène deux conséquences : premièrement, lorsqu'on suppose que l'échantillonneur est fermé, l'air pénètre dans l'échantillonneur en contaminant l'adsorbant et diminue ainsi la capacité d'adsorption. En deuxième lieu, lors des tests d'adsorption ou lors de l'étape de désorption, les fuites occasionnent des remplissages incomplets des sacs de collecte des gaz. Les volumes sont alors plus petits que les volumes théoriques attendus, ayant par effet de surestimer les émissions réelles. Les fuites provenaient de la zone d'union de la cartouche centrale avec les cônes et, dans quelques cas, des écrous aux extrémités des échantillonneurs.
1.4.4.4 Difficultés de manipulation
Deux inconvénients à la manipulation de l'assemblage de l'échantillonneur sont signalés. Premièrement, les cônes de l'échantillonneur doivent être serrés fermement à l'aide d'un marteau et d'un outil adapté à cette fin dans le but de rendre l'échantillonneur étanche. Cette pratique peut occasionner des dommages à l'appareil ou même un accident à l'opérateur. De plus, cette pratique est parfois inefficace et l'échantillonneur peut présenter encore des fuites. Deuxièmement, l'outil qui est utilisé afin de serrer les anneaux de soutien dans la cartouche glisse facilement et abime les surfaces latérales de la cartouche. En conséquence, les stries générées deviennent des chemins par lesquelles les gaz peuvent s'échapper.
2. Hypothèses, but et objectifs
Le but de la présente recherche est d'optimiser l'échantillonneur à flux passif développé par Godbout et al. (2006) et Gaudet (2005) et d'améliorer ses performances. À ce propos, cette étude est divisée en quatre volets.
Le premier volet a comme objectif l'étude et la mise en œuvre d'une série de modifications à apporter à l'échantillonneur lui-même (étanchéisation, retrait du diffuseur et remplacement de la plaque à orifice) et aux procédures d'utilisation (remplacement de l'azote par l'hélium, purgation des conduits et installation de valves) afin d'en améliorer la performance et l'assemblage. La performance de l'échantillonneur fait référence à son efficacité d'adsorption, son efficacité de désorption, sa précision et son exactitude pour mesurer les flux de N2O. L'hypothèse à vérifier est : « Les modifications apportées à l'échantillonneur et aux modes opératoires augmentent la performance par rapport à celle du modèle de Godbout et al. (2006) et Gaudet (2005) ».
Dans le deuxième volet, des modifications sont proposées à la cartouche afin d'augmenter la capacité d'adsorption du N2O de l'échantillonneur. L'objectif est d'analyser l'influence de l'augmentation de la capacité de la cartouche sur les performances de l'échantillonneur. Pour ce volet, l'hypothèse à vérifier est : « Le système permettant l'augmentation de la capacité d'adsorption ne réduit pas la performance des échantillonneurs ».
Le troisième volet a comme objectif de proposer une nouvelle procédure de désorption afin d'augmenter l'efficacité de désorption et d'améliorer ainsi la performance de l'échantillonneur. L'hypothèse à vérifier est : « La procédure de désorption proposée avec une température de 200 °C, un débit de 42 ml min"1 et un temps de désorption de 15
minutes augmente la performance de l'échantillonneur ».
Finalement, le quatrième volet est consacré à la calibration des échantillonneurs. L'objectif est de déterminer les constantes de calibration et leur linéarité pour chaque échantillonneur. L'hypothèse à vérifier est : « Les constantes de calibration des échantillonneurs modifiées conservent leur linéarité ».
3. Méthodologie
3.1 Volet 1 : Amélioration de la performance de l'échantillonneur
3.1.1 Étanchéisation de l'échantillonneur
Les fuites de gaz de l'échantillonneur développé par Godbout et al. (2006) et Gaudet (2005) proviennent du joint entre la cartouche et les cônes. De plus, quelques-uns des échantillonneurs présentent des fuites aux extrémités des cônes, que ce soit dans le tuyau d'entrée ou dans le tuyau de sortie.
Afin de résoudre le problème des fuites de la cartouche, différentes techniques d'étanchéisation ont été essayées. L'évaluation de chaque technique prend en considération sa capacité à supporter des températures de 350 °C et la présence de fuites. Cette température correspond à la température de conditionnement de la zéolite. La présence de fuites est vérifiée au moyen de l'application du liquide de détection de fuites Snoop® lorsque les échantillonneurs sont soumis à une pression interne de 200 kPa. La première technique utilise l'application du scellant de marque MASTERS, un composé métallique pour le scellement de filets de conduite de métal ou de plastique. Il est appliqué sur les filets autour des cartouches (figure 7A).
Pour la seconde technique, des rondelles plates en cuivre (30 mm de diamètre intérieur, 38 mm de diamètre extérieur et 0,16 mm d'épaisseur) sont installées entre la cartouche et les cônes (figure 7B). Afin d'éviter les fuites causées par des rayures sur les pièces, les surfaces des rondelles et celles de l'échantillonneur sont polies avec un papier sablé à l'eau très fin (P800). Une attention spéciale doit être accordée à ces pièces au cours de l'assemblage de l'échantillonneur en évitant le contact des surfaces avec les outils métalliques et en les déposant dans des contenants à surface douce.
La troisième et dernière technique utilise des rondelles en Viton de 34,93 mm de diamètre et 3,18 mm d'épaisseur (figure 7C). Elles sont placées entre la cartouche et les cônes. Le Viton est un fluor-élastomère possédant une large résistance chimique et une bonne résistance aux fluctuations de température jusqu'à un maximum de 315 °C.
B
r\
oo
Figure 7. Techniques d'étanchéisation, A : application d'un scellant de filets de conduite, B : installation de rondelles plates en cuivre et C : installation de rondelles en Viton.
Suite aux tests, l'application du scellant sur les filets des échantillonneurs n'a pas présenté une bonne tolérance aux températures soumises (300 à 350 °C), car le composant scellant a réagit à la chaleur et a laissé des résidus brûlés qui se collaient autour des filets. Lors de l'inspection des fuites, celles-ci étaient encore présentes. L'utilisation de cette technique a alors été rejetée.
Les rondelles en cuivre installées entre la cartouche et les cônes n'ont pas présenté d'altérations par les températures soumises (300 à 350 °C). En effet, le cuivre peut supporter des températures beaucoup plus élevées. Cependant, les échantillonneurs ont présenté parfois des fuites de gaz et pour cette raison, l'application de cette technique a été rejetée. Elle pouvait facilement occasionner des dommages aux échantillonneurs puisque ceux-ci devaient être serrés à l'aide d'un marteau et d'un outil spécifique à cette fin.
Lors de l'étape du conditionnement des échantillonneurs, les rondelles en Viton ont montré à l'occasion de légères altérations et dommages, surtout quand ils étaient trop serrés dans les échantillonneurs. Selon les spécifications fournies par le fabriquant, le Viton résiste à des fluctuations de température allant jusqu'à un maximum de 315 °C. Alors, de nouveaux tests ont été réalisés avec des échantillonneurs serrés à la main sans trop d'effort et soumis à une température de conditionnement de 310 °C. De cette façon, l'inspection n'a détecté aucune fuite et les anneaux n'ont pas présenté de détériorations causées par la température. Les mêmes résultats sont obtenus même après huit tests sans remplacement des anneaux. Cette technique a été adoptée parce que, en plus de rendre les échantillonneurs étanches, elle permet de réaliser leur assemblage et leur désassemblage d'une façon très pratique et efficace. Le schéma des toutes les pièces formant le nouvel échantillonneur est présenté dans l'annexe A.
Afin de pallier aux problèmes de fuites aux extrémités des cônes, les tuyaux à la sortie ou à l'entrée des échantillonneurs qui présentaient ce problème ont été remplacés. Suite à ces modifications des échantillonneurs complètement étanches ont été obtenus.
3.1.2 Retrait du diffuseur
La simulation de la dynamique de l'air à l'intérieur de l'échantillonneur faite par Gaudet (2005) avant de construire son prototype a montré que l'air arrive à une vitesse plus élevée sur les deux tiers centraux de la surface de l'adsorbant que sur l'autre tiers. C'est pourquoi, un diffuseur conique qui dirige l'air vers l'adsorbant à une vitesse constante sur toute la surface avait été conçu.
À l'aide du logiciel FLUENT, une nouvelle simulation de la dynamique de l'air à l'intérieur de l'échantillonneur a été refaite. Les valeurs de vitesse d'entrée d'air simulées ont été prises à partir des résultats des expériences réalisées par Gaudet (2005) lors de la détermination des constantes de calibration (K). La figure 8 permet de voir la simulation de la vitesse de l'air à l'intérieure de l'échantillonneur lorsque la vitesse de l'air à l'extérieur est de 10 m s . Si l'échantillonneur a une constante de calibration K égale à 0,0038 (Gaudet 2005), la vitesse d'entrée de l'air serait de 0,04 m s"1, ce que diffère
considérablement des valeurs utilisées par Gaudet (2005) (autour de 3 m s"1).
Figure 8. Vecteurs de vitesse de l'air dans l'échantillonneur colorés par magnitude (m s"1) et
Le volume de contrôle présenté à la figure 8 représente l'aire axisymétrique à l'axe X du cône antérieur de l'échantillonneur.
À cet égard, la simulation permet d'observer que l'air se répand dans le cône et arrive avec une vitesse uniforme sur toute la surface de l'adsorbant. Cette propagation de l'air dans l'échantillonneur est causée par les efforts visqueux qui dominent la dynamique de un écoulement à très basse vitesse. Pour cette raison, l'utilisation du diffuseur n'est plus nécessaire et il a été retiré de la cartouche.
Comme le diffuseur est attaché par des vis qui sont placées au milieu du matériel adsorbant, le retrait du diffuseur permet d'augmenter de 13 % la quantité d'adsorbant dans la cartouche.
3.1.3 Réduction de l'excès de N20
Dans les résultats obtenus par Gaudet (2005), la quantité de N20 récupérée à la
désorption est toujours plus grande que la quantité établie dans chaque expérience (en moyenne 34 % de N2O de plus). La mauvaise étanchéité de l'échantillonneur est une des raisons qui explique ce phénomène, comme il a été mentionné auparavant. Gaudet (2005) affirme que la cause de cet excès est attribuée au N2O provenant de l'air ambiant qui se trouve dans les tuyaux des montages avant l'exécution de chaque étape.
Dans le but de pallier à cet excès de N2O attribué à l'air dans les tuyaux des montages, les mesures suivantes ont été adoptées :
1. Gaudet (2005) emploie de l'azote (N2) pour réaliser le conditionnement de l'adsorbant et sa désorption. Puisque le gaz ciblé est un composé azoté, le N2 a été remplacé par l'hélium (He) afin d'éviter de possibles réactions entre le N2, le N2O et l'air ambiant.
2. Lors de la construction des montages, les tuyaux ont été coupés à la longueur minimale.
4. Des valves ont été installées sur chaque ligne de conduite de gaz afin de restreindre l'entrée d'air une fois les tuyaux purgés.
3.1.4 Installation de la plaque à orifice
Comme le montre la figure 5 à la section 1.4, une plaque à orifice est installée à la sortie de l'échantillonneur. Cette plaque est si petite qu'elle peut tomber et s'égarer facilement lors du montage étant donnée la façon dont elle est placée. La perte de cette pièce lors d'une séance d'échantillonnage sur le terrain peut entraîner des coûts supplémentaires, une perte de temps et l'invalidation des résultats. Pour cette raison, les plaques à orifice ont été fixées dans des raccords qui sont placés à la sortie de chaque échantillonneur (figure 9). Un plan schématique général du prototype de l'échantillonneur modifié et de ses pièces est présenté à l'annexe A.
Figure 9. Plaque à orifice accouplée au raccord
3.1.5 Mode opératoire
3.1.5.1 Conditionnement de l'adsorbant
Le conditionnement a pour but le nettoyage de l'adsorbant et la préparation des échantillonneurs pour les séances d'adsorption. Le montage (figure 10) est constitué d'un cylindre d'hélium agissant comme gaz nettoyant inerte (figure 10-1), une valve contrôlant le flux de gaz (figure 10-2), un débitmètre (0 - 5 L min"1) (figure 10-3), un distributeur de
flux en acier inoxydable répartissant le gaz à chaque échantillonneur (figure 10-4) et un four capable d'atteindre une température de 310 °C (figure 10-5). Cette étude utilise
quatre échantillonneurs au total. L'écoulement de gaz s'effectue à l'intérieur de tuyaux d'acier inoxydable de 6,35 mm (1/4 po.).
Figure 10. Système de conditionnement des échantillonneurs
Voici la procédure :
1. Connecter les entrées des échantillonneurs au distributeur et les placer dans le four. Le four utilisé a la capacité de stocker quatre échantillonneurs simultanément. S'assurer de laisser l'autre extrémité des échantillonneurs libre pour permettre la sortie du gaz.
2. Chauffer le four à 310 °C.
3. Une fois la température atteinte, ouvrir la valve et injecter 600 ml min"1 d'hélium
par échantillonneur. S'il y a quatre échantillonneurs dans le four, ajuster le débit de gaz à 2400 ml min"1.
5. Immédiatement après les 180 min, fermer la valve, retirer les échantillonneurs du four et boucher leurs extrémités le plus rapidement possible afin d'éviter le contact de la zéolite avec l'air.
6. Une fois les échantillonneurs refroidis, ils sont prêts pour réaliser les mesures d'adsorption. Les bouchons ne doivent pas être retirés avant les expériences d'adsorption ou d'échantillonnage afin d'éviter que la zéolite n'adsorbe des gaz ambiants.
3.1.5.2 Protocole de l'expérience d'adsorption
Le but de cette expérience vise à déterminer l'efficacité d'adsorption de l'échantillonneur avec les nouvelles conditions et en même temps, de déterminer l'influence du débit de gaz sur l'efficacité de l'adsorption. Le montage présenté à la figure 11 est constitué d'une bonbonne de N2O de 2 ppm (figure 11-1), une valve micrométrique (figure 11-2), un débitmètre (figure 11-3), un échantillonneur à tester (figure 11-4), un raccord contenant la plaque à orifice (figure 11-5) et un sac de stockage du gaz traversant l'échantillonneur (figure 11-6).
Le test consiste à connecter l'échantillonneur directement à une source de N2O de concentration connue. Le montage permet de contrôler et mesurer le débit de gaz qui passe par l'échantillonneur. Au moment où le gaz traverse l'adsorbant, les molécules de N2O sont adsorbées. Par la suite, le gaz sortant de l'échantillonneur est collecté dans des sacs. La concentration de N2O contenue dans le sac est analysée au moyen d'un chromatographe en phase gazeuse (CG).
Avant d'installer l'échantillonneur et le sac collecteur sur le montage, l'air initialement présent dans les tuyaux du système est évacué afin d'éviter l'adsorption de composants autres que le N2O et, par conséquent, la contamination des échantillons. Ainsi, l'évacuation est réalisée, avant chaque session d'expériences, par l'injection d'un flux de N2O dans les tuyaux du système. Une fois l'air évacué, la valve est fermée pour éviter un retour de l'air.
Voici la procédure d'adsorption :
1. Connecter le raccord contenant la plaque à orifice au tuyau du sac collecteur. 2. Enlever le bouchon de la sortie de l'échantillonneur et connecter le raccord de la
plaque à orifice. Ensuite, enlever le bouchon de l'entrée de l'échantillonneur et connecter l'échantillonneur au débitmètre.
3. Ouvrir la valve du système et la valve du sac. Ajuster le débit désiré. La durée de chaque expérience d'adsorption est de 20 minutes.
4. Une fois les 20 minutes complétées, fermer la valve du sac et la valve du système. 5. Retirer l'échantillonneur du système et remettre les bouchons rapidement.
6. Déterminer la concentration de N2O du sac au moyen du CG.
7. La quantité de N2O adsorbé par la zéolite est déterminée à partir de la différence entre la masse de gaz envoyée à l'échantillonneur et la masse de gaz collectée dans le sac :
Où : m ^ = Quantité de N20 adsorbé par la zéolite, ng
mm a i e= Quantité totale de N20 injectée dans l'échantillonneur, ng
ms = Quantité N2O qui n'est pas adsorbé par la zéolite, ng
La masse est calculée à partir de l'équation suivante :
m = [N20]V pN i 0 [8]
Où : m = Masse de N2O, ng
[N20] = Concentration de N20, ppm
V = Volume de gaz dans le sac, ml
pN 0= Densité du N2O à température ambiante (1,8 mg ml"1)
8. Calculer l'efficacité d'adsorption. Elle est déterminée à partir du rapport entre la masse de N20 adsorbée et la masse totale de N2O qui a passé par
l'échantillonneur (équation 9).
( Wads ~ ma d s
V mtotale J
xlOO [9]
Où : 77^ = Efficacité d'adsorption de l'adsorbant dans l'échantillonneur, %
9. Répéter les étapes 1 à 7 pour les échantillonneurs restants. L'ordre des essais est réalisé en suivant le plan expérimental présenté à la section 3.1.3.
Les sacs, d'une capacité de 2 L, sont conditionnés après chaque analyse afin de les réutiliser. La procédure de conditionnement des sacs consiste à expulser le N2O de l'intérieur et à les nettoyer afin d'assurer l'absence de traces de N2O. Lors du nettoyage, les sacs sont remplis d'hélium et ensuite vidés complètement à nouveau à l'aide d'une pompe à vide. Cette procédure est répétée deux fois pour chaque sac après chaque utilisation.
Dans le cadre de la validation de l'échantillonneur, l'efficacité d'adsorption doit être indépendante de la vitesse du gaz qui traverse l'adsorbant. Pour vérifier cette condition, une analyse statistique est effectuée où l'hypothèse posée est la suivante :
H0 : Les efficacités d'adsorption sont égales.
3.1.5.3 Protocole de l'expérience de désorption
La désorption est le procédé d'extraction et de collecte du gaz adsorbé par la zéolite. Le gaz est collecté dans des sacs et quantifié au moyen du CG. La désorption utilisée est de type thermique, ce qui implique l'utilisation de la chaleur et un écoulement de gaz inerte à travers l'adsorbant afin de volatiliser le N2O, le dégager de la zéolite et le conduire vers le sac collecteur.
Le montage construit a la capacité de désorber et collecter le N2O de quatre échantillonneurs simultanément. Des débitmètres et des valves micrométriques ont été installés sur chaque ligne de conduite de gaz afin d'augmenter la précision des mesures. La figure 12 présente le montage et ses composants : un cylindre d'hélium de grade 5.0 (figure 12-1), quatre valves micrométriques qui ajustent le débit du gaz de chaque ligne (Figure 12-2), quatre débitmètres (0-100 ml min"1) (figure 12-3), un four capable
La procédure de désorption suivie consiste à :
1. De façon identique à la procédure d'adsorption, l'air qui est initialement présent dans les tuyaux du système est évacué avant chaque session de désorption. L'évacuation est réalisée par l'injection d'hélium dans le système avant d'installer l'échantillonneur. Une fois l'air évacué, les valves sont fermées pour éviter l'entrée d'air.
2. Enlever les bouchons et installer l'échantillonneur à une ligne de conduite de gaz dans le four. Répéter cette étape avec les échantillonneurs restants.
3. Chauffer le four à 100 °C.
4. Lorsque le four atteint 100 °C, ouvrir les valves des sacs et régler les valves micrométriques afin d'obtenir un débit de 21 ml min"1. Mettre en marche le
chronomètre.
5. Après 30 minutes, fermer les valves des sacs et les valves micrométriques. 6. Déterminer la concentration de N2O des sacs au moyen du chromatographe. 7. Déterminer la masse de gaz dans chaque sac selon l'équation suivante :
mdes=[N20}V pN i 0 [10]
Où : mdes = Quantité de N2O désorbée, ng
[N20] = Concentration du N2O dans le sac déterminée par le CG, ppm
V = Volume de gaz dans le sac de désorption, ml pN O = Densité du N2O à température ambiante, mg ml"1
8. Finalement, calculer l'efficacité de désorption :
xlOO [11]
f... \
Vdes =
™des \ma d s )
3.1.6 Dispositif expérimental
Le plan de l'expérience est structuré comme un « carré latin » de 4x4. Quatre échantillonneurs ont été identifiés par une lettre inscrite sur chaque pièce, soit : C, D, E et Y. En tenant compte de la recommandation faite par Gaudet (2005) d'évaluer l'efficacité d'adsorption de l'échantillonneur avec des débits correspondant à des vitesses d'air extérieur plus grands que 7 m s"1, au total, quatre débits de N2O sont analysés (6,5; 13;
19,5 et 26 ml min"1).
Généralement, à l'intérieur d'une journée, il est possible de réaliser une séance de quatre essais. Par conséquent, les blocs formés correspondent aux jours d'essais et les traitements correspondent aux quatre débits évalués. En formant quatre blocs dans lesquels un débit est assigné à chacun des échantillonneurs de façon aléatoire, les traitements sont équilibrés en permettant que chaque échantillonneur soit testé un nombre égal de fois avec chaque débit (tableau 2). Puisque le débit d'adsorption n'influence pas la performance de l'échantillonneur à la désorption, l'effet nommé « N2O injecté » est ajusté avec l'effet aléatoire des blocs dans l'analyse statistique comprise dans un modèle mixte. Les blocs et les traitements sont assignés selon la procédure de randomisation pour un carré latin 4x4 à l'aide de la fonction ALEA de Microsoft Excel.
Tableau 2. Plan expérimental pour l'étude de la performance de l'échantillonneur
Débit1 N20 injecté (ng) Bloc (ml min'1) N20 injecté (ng) 1 2 3 4 6,5 0,468 Y* E C D 13 0,936 C Y D E 19,5 1,404 D C E Y 26 1,872 E D Y C
1 : débit de N20 à l'adsorption, ml min" ; 2 : identification de l'échantillonneur
Chaque essai d'adsorption est d'une durée de 20 minutes afin de ne pas saturer l'adsorbant. Le volume de gaz collecté à l'adsorption est de 130, 260, 390 et 520 ml pour des débits de 6,5, 13, 19,5 et 26 ml min"1 respectivement. Le temps d'adsorption et la
( = _ C A T M _
Q[N20]pN i 0
CATNi0=mzCAUNi0 [13]
Où : t = Temps d'adsorption avant la saturation de la zéolite, min CATNO = Capacité d'adsorption totale de N2O de l'adsorbant, ng
Q = Débit de gaz qui passe par l'adsorbant, ml min"1
mz = Quantité d'adsorbant dans l'échantillonneur, (749 mg)
CAUNO = Capacité d'adsorption unitaire de N2O de l'adsorbant,
(3,96 ngN2o mg'zéoiite) (Godbout et al. 2006)
3.1.7 Exactitude et précision de l'échantillonneur
L'ANSI/ASHRAE (1992) définit l'erreur comme la différence entre la vraie valeur de la quantité mesurée et la valeur observée. L'erreur expérimentale peut être de deux types : erreur systématique (exactitude) ou erreur aléatoire (précision).
L'erreur systématique est récurrente et ne peut être uniquement expliquée que par le hasard. Une erreur de ce type peut, par exemple, être corrigée par la calibration (ANSI/ASHRAE, 1992). Ainsi, l'exactitude de l'échantillonneur est déterminée à partir de l'efficacité totale de ce dernier. En effet, il s'agit du ratio entre la quantité de N2O désorbée et mesurée par le CG et la quantité total de N2O injectée dans le système et dirigée vers l'échantillonneur :
m
»7«*=^
I totale 2-xl00 W
m totale
Où : rjtolale = Exactitude ou efficacité totale de l'échantillonneur, % mdes = Quantité de N20 désorbé, ng
L'erreur aléatoire est une lecture erronée qui apparaît avec régularité de chaque coté d'une certaine valeur moyenne. Les mesures peuvent être précises ou imprécises selon la capacité de l'instrument à reproduire avec régularité de lectures subséquentes (ANSI/ASHRAE, 1992). La précision de l'échantillonneur indique la dispersion des mesures ou la variabilité de la méthode. Dans cette étude, le coefficient de variation est le paramètre statistique de dispersion employé pour faire référence à la précision de mesure de l'échantillonneur. Donc, à partir des résultats de l'efficacité totale de l'équation [14], le coefficient de variation est calculé avec les équations [15] et [16]. De la même manière, la précision de l'adsorption de l'échantillonneur et de l'étape de désorption peut être calculée avec ces équations.
[15]
Y.
G -C V = = xlOO x 1 " « - 1 1 ^ - ~ x )2 [16]Où : CV = Coefficient de variation, % a = Écart type, %
x= Moyenne de l'efficacité totale, d'adsorption ou de désorption, ng xi = Efficacité totale, d'adsorption ou de désorption de chaque essai, %
n = Nombre d'essais ou observations
3.2 Volet 2 : Augmentation de la capacité d'adsorption
3.2.1 Augmentation de la capacité de stockage de la zéolite dans l'échantillonneur
Dans le but d'augmenter la capacité d'adsorption du N2O de l'échantillonneur, il est proposé d'augmenter la quantité d'adsorbant dans la cartouche. Godbout et al. (2006) ont démontré que la capacité d'adsorption du N2O par la zéolite 5A dépend linéairement de la quantité présente dans l'échantillonneur. À cet égard, un système permettant d'augmenter
la capacité d'adsorbant dans la cartouche a été développé et son influence sur la performance de l'échantillonneur a été étudiée.
L'échantillonneur modifié présenté à la section 3.1 est capable de stocker une couche de 749 mg de zéolite 5A d'une épaisseur de 2 mm. Afin d'augmenter cette capacité, la conception des grillages qui retiennent la zéolite a été modifiée pour obtenir une couche plus épaisse d'adsorbant. Au total, trois épaisseurs additionnelles ont été développées, soit 4, 6 et 8 mm, ce qui a permis de stocker respectivement 1467, 2153 et 2810 mg de zéolite. Les nouvelles épaisseurs représentent une augmentation de 96, 187 et 275 % de la capacité initiale d'adsorption.
La figure 13 présente le schéma des grillages constitués de deux pièces. La première pièce, dans laquelle la zéolite est stockée, a la forme d'un récipient (figure 13-1). Elle est placée en aval de la cartouche. Sa profondeur détermine l'épaisseur de la couche. La deuxième pièce agit comme un couvercle afin de retenir la zéolite (figure 13-2). Elle est placée à l'arrière de la cartouche. Les nouveaux grillages utilisent les mêmes anneaux de soutien que la conception originale de l'échantillonneur.
2mm
"Li:
4mmr~\ J - /—^
T
t
_^-25 mm 30 mm # Aire effective O Aire de support3.2.2 Mode opératoire
Les étapes de conditionnement, d'adsorption et de désorption pour l'étude de la performance de l'échantillonneur avec les nouvelles épaisseurs de zéolite, suivent les modes opératoires présentées à la section 3.1.5. Cependant, les essais sont réalisés avec le plan expérimental présenté à la section.3.2.3.
3.2.3 Plan expérimental
La performance de l'échantillonneur avec chacune des épaisseurs d'adsorbant (2, 4, 6 et 8 mm) est évaluée pour quatre débits de gaz contenant 2 ppm de N2O, soit 6,5, 13, 19,5 et 26 ml min"1 (tableau 3). Le temps d'adsorption est fixé à 20 minutes par traitement,
représentant une masse totale injectée respectivement de 0,468, 0,936, 1,404 et 1,872 .ug de N20.
Tableau 3. Plan expérimental pour l'étude de l'influence de l'augmentation de la quantité de zéolite sur la performance de l'échantillonneur
Bloc Essai Éch.1 Couche N20 total2
# (mm) injecté (ng) 1 D 6 1,872 1 2 C 8 0,936 3 E 4 1,404 4 Y 2 1,404 5 Y 2 0,936 2 6 C 8 1,404 2 7 D 6 1,404 8 E 4 0,936 9 C 8 1,872 3 10 D 6 0,936 3 11 Y 4 0,468 12 E 4 1,872 13 E 2 1,872 4 14 C 8 0,468 4 15 D 6 0,468 16 Y 2 0,468
Bloc Essai Éch.1 Couche N20 total2
# (mm) injecté (ng) 17 D 6 0,468 5 18 Y 4 1,404 19 C 4 0,468 20 E 6 1,404 21 E 8 0,468 6 22 D 8 0,936 6 23 Y 6 0,936 24 C 8 1,404 25 D 2 0,468 7 26 E 2 1,404 27 C 4 1,872 28 Y 4 0,936 39 E 2 1,872 8 30 C 6 1,872 8 31 Y 8 1,872 32 D 2 0,936
1 Échantillonneur : C, D, E ou Y;2 Cette valeur représente la masse totale de N20 traversant