pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS (Faculté des Sciences Fondamentales et Appliquées)
(Diplôme National - Arrêté du 7 août 2006)
Ecole Doctorale : Sciences pour l’Environnement Gay Lussac N°523 Secteur de Recherche : Chimie organique, minérale et industrielle
Présentée par :
Maxime PHILIPPE
Ingénieur ENSCCF
************************
IMPACT DES COMPOSES OXYGENES MODELES ISSUS
D’UNE HUILE DE PYROLYSE DE BIOMASSE SUR
L’HYDRODESULFURATION DES DIBENZOTHIOPHENES
************************
Directeur de Thèse : Sylvette BRUNET, Chargée de Recherche CNRS, LACCO Poitiers Co-directeur de Thèse : Frédéric RICHARD, Maître de Conférences, LACCO Poitiers
************************ Soutenance prévue le 26 octobre 2010
devant la Commission d’Examen
************************
JURY
Rapporteurs : Christophe GEANTET, Directeur de Recherche CNRS, IRCELYON Villeurbanne Edmond PAYEN, Professeur, UCCS Lille
Examinateurs : Sylvette BRUNET, Chargée de Recherche CNRS, LACCO Poitiers Daniel DUPREZ, Directeur de Recherche CNRS, LACCO Poitiers Bruno GAGNEPAIN , Ingénieur de Recherche, ADEME Angers
Damien HUDEBINE , Ingénieur de Recherche, IFP Energies nouvelles Lyon Frédéric RICHARD, Maître de Conférences, LACCO Poitiers
INTRODUCTION GENERALE ... 1
CHAPITRE I : ETUDE BIBLIOGRAPHIQUE ... 7
I - Les catalyseurs d’hydrotraitement ... 9
II - hydrotraitement des gazoles... 15
II. 1 - Composition globale d'un gazole ... 15
II. 2 - L'hydrotraitement catalytique... 15
II. 2. 1 - Hydrodésulfuration de molécules soufrées modèles ... 17
II. 2. 2 - Effet de la pression partielle en sulfure d’hydrogène lors de l'HDS des molécules soufrées réfractaires... 20
III - Procédés d’hydrodésoxygénation (HDO) ... 21
III. 1 - Obtention et valorisation de coupes issues de la biomasse ligno-cellulosique... 22
III. 2 - Procédé d’hydrodésoxygénation des huiles brutes ... 25
III. 3 - Transformation de charges modèles de bio-huiles... 26
III. 4 - Transformation de différentes molécules modèles ... 28
III. 4. 1 - Le benzofurane... 28
III. 4. 2 - L’heptanoate de méthyle ... 29
III. 4. 3 - Les composés phénoliques ... 31
III. 4. 4 - Transformation du CO – Réaction de water gas shift ... 34
III. 5 - Influence des sous-produits de réaction sur l’HDO de molécules oxygénées ... 36
III. 6 - Interaction entre composés oxygénés et la surface des catalyseurs ... 38
IV - Hydrodésoxygénation et hydrodésulfuration simultanées... 39
V - Conclusion... 41
CHAPITRE II : PARTIE EXPERIMENTALE... 43
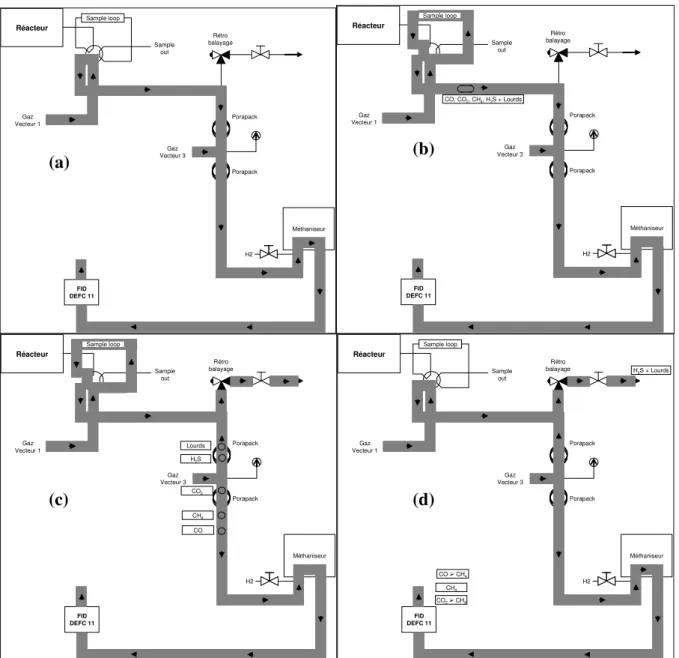
I - Appareillage ... 45
II - Conditions opératoires... 47
II. 1 - Catalyseur... 47
II. 1. 1 - Chargement du réacteur... 47
II. 1. 2 - Sulfuration ... 48
II. 3. 1 - Analyse des gaz ... 51
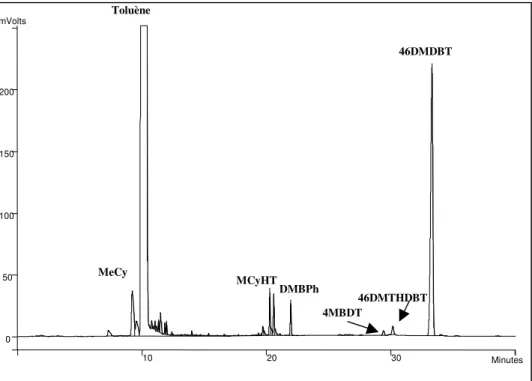
II. 3. 2 - Analyse des produits organiques liquides... 54
III - Exploitation des résultats... 61
III. 1 - Conversion et rendements molaires pour la transformation des composés soufrés (DBT ou 46DMDBT)... 61
III. 2 - Conversion, rendements molaires et bilans molaires pour la transformation des composés oxygénés (guaiacol, phénol ou acide décanoïque) ... 62
III. 3 - Sélectivité et activité catalytique ... 63
IV - Caractérisation du catalyseur après réaction... 65
IV. 1 - Analyse des teneurs en soufre et en carbone ... 65
IV. 2 - Spectroscopie de photoélectrons X... 65
V - Produits chimiques utilisés ... 68
CHAPITRE III : TRANSFORMATION DES MOLECULES MODELES SEULES .... 69
I - Hydrodésulfuration du DBT et du 46DMDBT ... 71
I. 1 - Schéma réactionnel du DBT et du 46DMDBT ... 72
I. 2 - Effet de la nature du solvant sur la transformation des composés soufrées ... 74
I. 3 - Comparaison des réactivités du 46DMDBT et du DBT... 75
II - Hydrodésoxygénation du guaiacol et du phénol... 77
III - Transformation du CO et du CO2... 80
III. 1 - Aspects thermodynamiques ... 80
III. 2 - Transformation du CO et du CO2 en absence et en présence de CoMoP ... 81
IV - Hydrodésoxygénation de l’acide décanoïque... 84
V - Discussion ... 88
VI - Conclusion ... 92
CHAPITRE IV : EFFET DES MOLECULES OXYGENEES SUR L’HYDRODESULFURATION DES DIBENZOTHIOPHENES... 95
I - Protocole expérimental... 97
II - Impact des molécules oxygénées sur l’HDS du DBT et du 46DMDBT... 99
conversions... 99
II. 1. 2 - Impact du guaiacol et du phénol sur l’HDS du DBT et du 46DMDBT à fortes conversions... 101
II. 1. 3 - Impact des molécules modèles soufrées sur l’HDO du guaiacol et du phénol ... 103
II. 2 - Impact du CO et du CO2 sur l’HDS du DBT et du 46DMDBT... 104
II. 2. 1 - Impact du CO et du CO2 sur l’HDS du DBT et du 46DMDBT à faibles conversions... 104
II. 2. 2 - Impact du CO et du CO2 sur l’HDS du DBT et du 46DMDBT à fortes conversions... 108
II. 2. 3 - Impact des molécules modèles soufrées sur la transformation du CO et du CO2... 108
II. 3 - Impact de l’acide décanoïque sur l’HDS du DBT et du 46DMDBT ... 109
II. 3. 1 - Impact de l’acide décanoïque sur l’HDS du DBT et du 46DMDBT à faibles conversions... 109
II. 3. 2 - Impact du DBT et du 46DMDBT sur la transformation de l’acide décanoïque ... 111
II. 3. 3 - Comparaison de l’effet inhibiteur du CO et de l’acide décanoïque sur l’HDS du DBT et du 46DMDBT... 113
II. 3. 4 - Impact de l’acide décanoïque sur l’HDS du DBT et du 46DMDBT à fortes conversions... 114
II. 4 - Modélisation de l’effet des molécules oxygénées sur l’HDS du DBT et du 46DMDBT ... 115
III - Modification des propriétés catalytiques du catalyseur CoMoP/Al2O3 après l’HDS de composés soufrés en présence des molécules oxygénées ... 120
III. 1 - Effet du guaiacol et du phénol ... 120
III. 2 - Effet du CO et du CO2... 124
III. 3 - Effet de l’acide décanoïque... 129
IV - Discussion ... 131
V - Conclusion... 135
CONCLUSION GENERALE ... 137
Le développement de procédés de production de carburant à partir de sources hydrocarbonées renouvelables (biomasse) est un axe de recherche important depuis ces dernières années. En effet, l'augmentation du prix du pétrole, la raréfaction des bruts et une demande sociétale de plus en plus poussée en termes environnementaux montrent que le seul pétrole ne pourra satisfaire les demandes énergétiques du futur.
Une réponse possible à ce nouveau défi consiste à proposer l'utilisation de la biomasse comme alternative écologique et durable au pétrole dans le domaine des carburants et plus particulièrement la fraction lignocellulosique des plantes (bois, paille, etc...) qui n'est actuellement pas valorisée sous forme de carburants.
Une des principales difficultés pour valoriser la matière lignocellulosique est sa forme solide qui la rend difficile à transporter de son lieu de prélèvement (forêts, champs, littoraux, etc...) vers son lieu d'utilisation (usines, villes, etc...). L'autre difficulté est que les moteurs actuels fonctionnent avec des carburants liquides et non solides et qu'il n'est pas économiquement envisageable de modifier les circuits de distribution en carburants. Il est donc nécessaire d'opérer une gazéification ou une pyrolyse de manière à transformer respectivement la matière lignocellulosique solide en gaz de synthèse ou en un liquéfiat. Le gaz de synthèse peut alors être converti dans un procédé de type Fischer-Tropsch de manière à obtenir des carburants de haute qualité. Les liquéfiats sont, quant à eux, exploités pour le moment exclusivement comme combustibles car ils contiennent de fortes quantités d'oxygène (jusqu’à 45 % pds) sous forme d'eau libre mais aussi de composés oxygénés (présentant des fonctions multiples telles que les acides carboxyliques, les esters, les alcools, etc...) ce qui les rend particulièrement instables (réactions de polymérisation) et peu énergétiques.
Pour valoriser les liquéfiats de biomasse sous forme de carburants, il est donc essentiel d'effectuer un traitement de manière à éliminer tout ou partie de l'oxygène, ceci de manière à les stabiliser et à les rendre compatibles avec les carburants pétroliers actuels. Pour cela, il peut être envisageable de développer de nouveaux procédés. Une solution plus simple, plus rapide et plus acceptable pour les industriels consisterait à ajouter aux gazoles conventionnels issus du raffinage une fraction assez faible de liquéfiats de biomasse avant de les envoyer dans un procédé classique d'hydrotraitement notamment d’hydrodésulfuration. De plus, il faut veiller à respecter les normes environnementales sur les teneurs en soufre dans les gazoles (10 ppm depuis 2009 en Europe par exemple) [1] puisque les composés oxygénés introduits peuvent être des inhibiteurs des réactions d’hydrodésulfuration. En effet, le soufre contribue de manière importante à l’empoisonnement irréversible des pièges à oxydes d’azote présents dans les pots catalytiques des véhicules automobiles et, dans une moindre mesure, à la
pollution atmosphérique par les rejets d’oxydes de soufre lors de la combustion dans le moteur. L'hydrodésoxygénation (HDO) se ferait alors en parallèle des réactions de désulfuration. L'avantage de cette solution est de ne pas modifier les schémas actuels de raffinerie permettant ainsi de limiter les investissements.
Très peu de travaux dans la littérature académique font état de ce type de procédés alors qu’il est indispensable de bien comprendre les phénomènes impliqués. En effet, la modélisation d’un tel procédé de co-traitement (hydrodésoxygénation - hydrodésulfuration) nécessite de connaître les mécanismes réactionnels détaillés de conversion des composés oxygénés ainsi que les interactions complexes qui peuvent exister entre composés soufrés et oxygénés, afin d’accéder aux cinétiques des réactions d’hydrodésulfuration (HDS) et d’hydrodésoxygénation (HDO). L’approche la plus simple est, dans un premier temps, est de développer une étude à partir de molécules modèles afin de simplifier les interactions entre les composés oxygénés et soufrés. Ceci nous permettera d’identifier les phénomènes impliqués de façon à acquérir les connaissances scientifiques indispensables à la modélisation des systèmes constitués par des charges réelles.
Dans ce contexte, les travaux effectués dans le cadre de cette thèse ont pour objectifs : 1/ d’étudier la transformation des molécules modèles oxygénées seules dans des conditions expérimentales proches de celles de l’HDS des gazoles en présence d’un catalyseur conventionnel CoMoP/Al2O3 (340°C, 4,0 MPa) afin de comparer leur réactivité et de
déterminer leur schéma de transformation.
Les composés oxygénés modèles choisis, représentatifs de ceux présents dans les liquéfiats de biomasse, sont d’une part le guaiacol et le phénol (principal produit de transformation du guaiacol) qui peuvent être considérés comme les composés oxygénés les plus réfractaires à l’hydrodésoxygénation, et d’autre part, l’acide décanoïque et ses sous-produits de décomposition (CO et CO2).
2/ de mettre en évidence l’impact de ces différents composés modèles oxygénés sur la transformation de deux composés modèles soufrés qui sont le dibenzothiophène (DBT) et le 4,6-diméthyldibenzothiophène (46DMDBT). En effet, ceux-ci sont parmi les composés les plus réfractaires à l’HDS et peuvent être considérés comme représentatifs de ceux contenus dans des coupes gazoles pour des teneurs en soufre inférieures à 250 ppm. En outre, la transformation de ces deux composés soufrés implique les deux principales voies de désulfuration qui sont la rupture directe des liaisons C-S (voie DSD, majoritaire pour la
transformation du DBT) et l’hydrogénation du noyau aromatique suivie de la rupture des liaisons C-S (voie HYD, majoritaire pour la transformation du 46DMDBT).
Les teneurs en composés oxygénés injectés sont comprises entre 1 et 5 % pds, ce qui correspond aux proportions pouvant être intégrées aux charges pétrolières.
Ce mémoire est divisé en quatre parties. Après avoir présenté les différents travaux de la littérature sur l’hydrotraitement de molécules modèles soufrées et oxygénées (Chapitre I), nous avons rappelé les techniques expérimentales et analytiques utilisées (Chapitre II). Dans le troisième chapitre, la transformation des molécules modèles soufrées et oxygénées seules est rapportée. Les schémas réactionnels des deux composés dibenzothiophéniques ont été confirmés et ceux des composés oxygénés établis dans les mêmes conditions opératoires. Enfin, dans le dernier chapitre, l’impact des molécules oxygénées sur la transformation des composés dibenzothiophéniques est présenté. Pour préciser l’origine des effets inhibiteurs et permettre leur quantification, une modélisation cinétique a été réalisée à partir des résultats expérimentaux obtenus lors de l’étude de l’effet des différents composés oxygénés (phénol, guaiacol, CO, CO2 et acide décanoïque) sur l’HDS des composés dibenzothiophéniques.
Enfin, le catalyseur CoMoP/Al2O3 a été caractérisé à chaque stade de la réaction (après
sulfuration et après le traitement des composés soufrés seuls et/ou en présence des composés oxygénés) afin de mettre en évidence les modifications éventuelles du catalyseur.
CHAPITRE I :
Cette partie résume l’état de l’art d’une part sur l’hydrodésulfuration (HDS) des gazoles et, plus spécifiquement sur l’HDS poussée des gazoles, et d’autre part sur l’hydrodésoxygénation (HDO) en général et plus précisément sur l’hydrodésoxygénation de molécules oxygénées modèles. Compte tenu du contexte de la thèse, les travaux en hydrodésulfuration et en hydrodésoxygénation simultanées sont tout spécialement présentés.
I - LES CATALYSEURS D’HYDROTRAITEMENT
Les catalyseurs classiques d’hydrotraitement sont généralement des catalyseurs supportés contenant des oxydes métalliques (des groupes VIB et VIII). Ils doivent donc être sulfurés pour être actifs en HDS et stables dans les conditions du procédé. Ces catalyseurs sont constitués de sulfure de molybdène (qui peuvent être promus par du nickel ou du cobalt) dont la structure et la morphologie ont fait l’objet de nombreuses analyses (Mössbauer, DFT, …) permettant d’améliorer la connaissance de ces matériaux.
Tout d’abord, la structure du catalyseur non promu (à base de sulfure de molybdène) est présentée. La stœchiométrie est alors proche de deux atomes de soufre pour un atome de molybdène (MoS2) [2]. Le sulfure de molybdène cristallise dans le système hexagonal avec
les cations M2+ au centre d'un prisme trigonal formé par 6 atomes de soufre S2-. Ces unités géométriques donnent naissance par assemblage à une structure en feuillets de type « SMoSSMoS ». Les feuillets de MoS2 n'interagissent alors entre eux que par des forces de
van der Waals.
On admet en général que les sites actifs sont des atomes de molybdène contenant des insaturations de coordination (appelés CUS : Coordinately Unsaturated Site) qui seraient situés en bordure des feuillets de MoS2, comme démontré par l'équipe de Tanaka [3] à partir
d’une approche fondamentale basée sur le concept de Siegel [4] pour les catalyseurs oxydes. Plusieurs types de sites pourraient être différenciés à la surface d'un catalyseur en fonction de leur degré de coordination. L'importance des lacunes anioniques (ou CUS) a également été mise en évidence par Kasztelan et al. [5] lors de travaux, notamment sur le
cis-penta-1,3-diène.
Daage et Chianelli [6] se sont intéressés à l'influence du taux d'empilement des feuillets sur la sélectivité de la réaction d'HDS pour l'hydrodésulfuration du dibenzothiophène sur des catalyseurs massiques MoS2. Un modèle nommé "Rim-Edge" a alors été proposé pour
Schéma I-1 : Modèle Rim-Edge proposé par Daage et Chianelli [6].
Ainsi, les bords des feuillets seraient actifs pour l'hydrogénation et l'hydrodésulfuration du dibenzothiophène, tandis que seule l'hydrogénation s'effectuerait sur les sites en bordure des feuillets situés aux extrémités des particules de MoS2. Une
modification de la sélectivité du catalyseur serait alors possible par la seule variation de l'empilement des feuillets.
Ces dernières années, les modélisations à partir de calculs DFT (Density Functional
Theory) réalisées par Raybaud et Coll. [7-10] et par Travert et al. [11] ont montré que la
morphologie des feuillets de MoS2 ainsi que la stabilité des sites de bords étaient sensibles
aux conditions de sulfuration et d’hydrotraitement, et plus particulièrement aux rapports des pressions partielles PH2S/PH2.
Ainsi, selon Lauritsen et al, avec un rapport molaire H2S/H2 de 500 [12,13], les
feuillets de MoS2 auraient une morphologie triangulaire alors qu’avec un rapport H2S/H2
proche de 0,1 (conditions classiques de sulfuration [10]), les feuillets de MoS2 auraient une
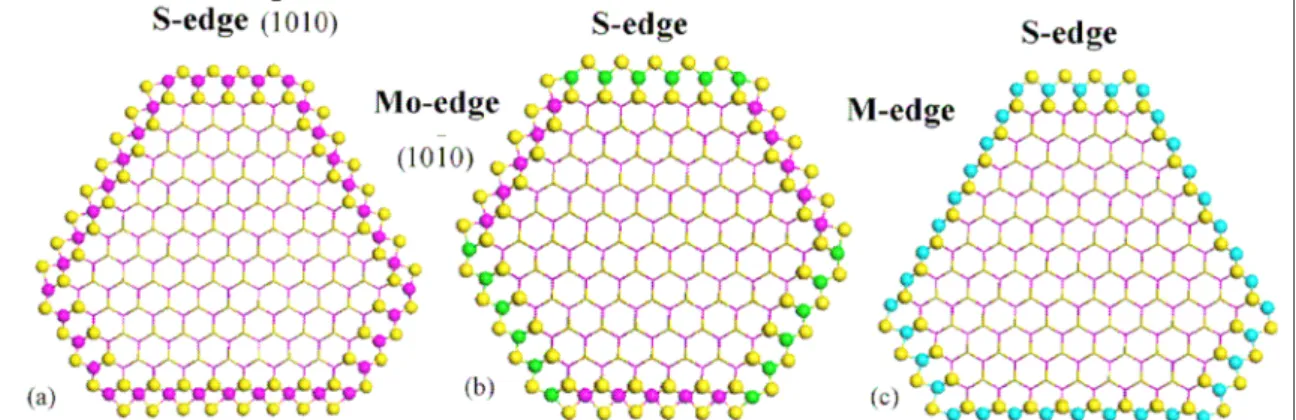
forme hexagonale tronquée. Lauritsen et al. [12] ont également observé ces différentes morphologies par Microscopie à Effet Tunnel (STM) et ont étudié des nanoclusters MoS2 de
forme triangulaire supportés sur une face (111) d’or, sous vide et sulfurés avec un rapport molaire H2S/H2 égal à 500 [13] (Figure I-1).
Lors de l'ajout de nickel ou de cobalt, un gain d'activité catalytique est observé correspondant à un maximum pour un rapport promoteur / (promoteur + molybdène) de l'ordre de 0,3 à 0,4 [14], par exemple pour l'HDS du dibenzothiophène (Figure I-2) :
Figure I-2 : Mise en évidence de l'effet promoteur du nickel (Ni) et du cobalt (Co) sur l'HDS du dibenzothiophène [14].
Au cours des années 70, plusieurs modèles [15-19] ont été développés pour rendre compte de cet effet promoteur du nickel ou du cobalt sur les catalyseurs. Cependant, le modèle de la "phase mixte" reste le plus largement admis à l'heure actuelle. Topsøe et al. [20] ont montré l'existence d'une phase mixte particulière (dénommée respectivement "CoMoS" ou "NiMoS" pour les catalyseurs supportés CoMo/Al2O3 et NiMo/Al2O3) à la surface du
catalyseur (Figure I-3). Cette phase posséderait une structure cristalline similaire à celle de MoS2, où les atomes de soufre monovalents liés au promoteur sont dans le plan du feuillet
MoS2. L’essentiel de l’activité catalytique en hydrotraitement est attribué à l’existence de
cette phase. Les atomes du promoteur, quant à eux, seraient situés aux arêtes des feuillets. Le promoteur (nickel ou cobalt) pourrait également être incorporé au support en substitution de l'alumine ou présent en surface du catalyseur sous forme de sulfure ou d'oxyde.
Plus récemment, Lauritsen et al. [13] ont observé par STM des feuillets de phase CoMoS et NiMoS (Figure I-4 et Figure I-5). Les feuillets de phases CoMoS adoptent une forme hexagonale presque régulière avec les atomes de cobalt localisés préférentiellement sur les bords soufre (1010) (Figure I-4). Les images STM des clusters de phase NiMoS révèlent que l’addition de nickel conduit à des morphologies tronquées où les bords métalliques (1010) sont majoritaires devant les bords soufre (1010). Ces auteurs [13] proposent que le nickel soit essentiellement localisé sur les bords soufre (Figure I-5).
Figure I-4 : (a) Image STM d’un feuillet CoMoS, (b) Modèle hexagonal de la phase CoMoS [13].
Figure I-5 : (a) Image STM d’un feuillet NiMoS, (b) Modèle hexagonal tronqué de la phase NiMoS [13]. Mo Co S Mo Ni S
Les études par microscopie à effet tunnel (STM) de Lauritsen et al. [12,13] sont en accord avec les travaux de Raybaud et Coll. (réalisés par calcul DFT) [7-10] puisque les deux groupes ont déterminé que dans les conditions usuelles d’hydrotraitement, les bords métalliques sont les plus abondants pour les catalyseurs promus. Concernant la localisation du cobalt, Lauritsen et al. [13] sont également en accord avec les conclusions obtenues par calcul DFT de Raybaud et Coll. [7,8] et de Paul et Coll. [11,21] puisque dans les conditions usuelles, le cobalt se substitue majoritairement au molybdène du bord soufre permettant aussi sa stabilisation. En revanche, la structure de la phase NiMoS proposée par Lauritsen et al. [13] avec du nickel substitué au molybdène uniquement sur le bord soufre est en désaccord avec de nombreuses études DFT [7,9,11,22] qui montrent que le nickel se repartirait de façon égale sur les deux types de bords.
Figure I-6 : Morphologie des phases sulfure obtenus par calcul DFT dans les conditions d’HDS pour (a) une phase MoS2, (b) une phase CoMoS et (c) une phase
NiMoS (jaune : soufre, magenta : molybdène, vert : cobalt, bleu : nickel) [10].
Raybaud et Coll. [8-10] ont pu simuler l’environnement des phases sulfures dans les conditions réelles d’HDS. La Figure I-6 montre les morphologies des phases sulfures (MoS2,
CoMoS et NiMoS) dans les conditions d’HDS (Tableau I-1). De plus, un rapport PH2S/PH2
élevé stabilise ce type de bord. Il est donc important de prendre en compte la nature de l’atmosphère entourant la surface étudiée dans le but d’en obtenir une description précise. Krebs et al. [23] ont établi des diagrammes à partir de calculs DFT permettant d’obtenir la morphologie du feuillet de CoMoS ou de NiMoS en fonction du potentiel chimique du soufre (Figure I-7). A 340°C et pour un rapport PH2S/PH2 de 0,042 (conditions utilisées lors de ce
travail de thèse), le potentiel chimique du soufre est de -0,9 eV. D’après les calculs réalisés par Gandubert et al. [24], la géométrie des feuillets de CoMoS serait hexagonale et peu déformée, le bord soufre serait composé de 100 % de cobalt avec une couverture en soufre de
50 % alors que le bord métallique n’exhiberait que 50 % de cobalt. La structure présentée Figure I-8 a été obtenue pour un rapport atomique Co/Mo de 0,29.
Tableau I-1 : Caractéristiques des phases sulfures déterminées par DFT [10].
Phase MoS2 CoMoS NiMoS
Forme Hexagonale
tronquée Hexagonale
Hexagonale fortement tronquée Emplacement du
promoteur - Bords soufre
Bords soufre et métalliques Couverture en soufre du bord métallique 100 % 100 % 0 % Couverture en soufre du bord soufre 50 % 50 % 50 %
Figure I-7 : Morphologie des feuillets de CoMoS (a) et de NiMoS (b) en fonction du potentiel chimique du soufre (∆∆∆∆µS) [23].
Figure I-8 : Représentation d’un feuillet de CoMoS (Co/Mo = 0,29) dans les conditions d’hydrotraitement (jaune : soufre, vert : molybdène, bleu : cobalt) [24].
Bord soufre Bord métallique
S
Mo
II - HYDROTRAITEMENT DES GAZOLES
II. 1 - Composition globale d'un gazole
Le gazole désigne une coupe d'hydrocarbures distillée en raffinerie dans une gamme de températures de distillation bien définie (entre 180 et 360 °C). Ainsi, on distingue trois familles principales de gazole (Tableau I-2) :
- le SRGO (Straight-Run Gas Oil) obtenu par la distillation atmosphérique du brut pétrolier, avec une teneur moyenne en soufre comprise entre 0,5 et 2,0 % poids et une teneur moyenne en azote de 50 à 500 ppm ;
- le LCO (Light Cycle Oil) provenant du craquage catalytique en lit fluidisé (Fluid
Catalytic Cracking ou FCC) à une concentration moyenne en soufre comprise entre 0,3 et 2,0
% poids et à une teneur moyenne en azote de 300 à 1000 ppm ;
- le CGO (Coker Gas Oil) issu du procédé de cokéfaction, dont les caractéristiques sont voisines de celles du SRGO en termes de soufre et d’aromatiques mais qui contient beaucoup plus de composés azotés (à des teneurs supérieures à 1000 ppm).
Tableau I-2 : Propriétés des gazoles SRGO, LCO et CGO [25].
Type de gazole SRGO LCO CGO
Origine distillation atmosphérique fraction distillée
du FCC Procédé de cokéfaction Température d'ébullition (°C) 180 - 360 180 - 360 180 – 360 10-40 % pds alcanes 10-30 % pds alcanes 30-70 % pds naphtènes 30-60 % pds naphtènes Composition 20-30 % pds aromatiques 60 à 90 % pds aromatiques 30-40 % pds aromatiques Indice de cétane 40 - 45 18 - 25 28 – 40 S (% pds) 0,5 – 2,0 0,3 – 2,0 1,5 – 3,0 N (% pds) 0,005 - 0,05 0,03 - 0,1 0,08 – 0,2
II. 2 - L'hydrotraitement catalytique
L’hydrotraitement est un procédé de raffinage qui consiste à éliminer les impuretés contenues dans les charges pétrolières telles que les composés contenant des hétéroatomes (S,
N et O) et des métaux (V, Ni) afin de préserver l'environnement ou d'éviter l'empoisonnement des catalyseurs utilisés dans d’autres procédés. C’est un procédé catalytique en lit fixe ou en lit bouillonnant sous pression d’hydrogène et pour des températures comprises entre 300 et 440°C. L’hydrotraitement permet aussi d’augmenter notablement la teneur en hydrogène des coupes pétrolières via l’hydrogénation des cycles aromatiques.
L'hydrotraitement intervient à différents niveaux dans les raffineries : en aval des procédés de raffinage du pétrole (pour éviter les rejets d'impuretés nuisibles à l'environnement lors de la combustion des carburants) ou en amont d'autres procédés, tels le reformage et le craquage, où les catalyseurs utilisés sont particulièrement sensibles à certains poisons comme le soufre et l’azote (Schéma I-2).
Selon l'hétéroatome à éliminer, on parle d'hydrodésulfuration ou HDS (pour le soufre), d'hydrodésoxygénation ou HDO (pour l'oxygène), d'hydrodésazotation ou HDN (pour l'azote) ou d'hydrodémétallation ou HDM (pour les métaux). L'hydrogénation des aromatiques (HDA) est considérée également comme une réaction d'hydrotraitement et consiste à hydrogéner les aromatiques pour améliorer par exemple l'indice de cétane du gazole, diminuer sa densité et ainsi respecter les spécifications imposées, les polyaromatiques contribuant à l'émission de composés toxiques et polluants par les moteurs Diesel.
Les catalyseurs industriels utilisés sont généralement des catalyseurs supportés dont la phase active est majoritairement du sulfure de molybdène promue par du nickel ou du cobalt. Les pressions totales d'utilisation sont comprises entre 10 et 250 bars en fonction de la sévérité de l’hydrotraitement à réaliser.
Nous développerons plus particulièrement dans la suite de ce mémoire les réactions d'hydrodésulfuration (HDS).
L'hydrodésulfuration (HDS) permet d'éliminer le soufre contenu dans les différentes coupes pétrolières par réaction sous pression d’hydrogène à haute température. Dans le cas des essences et des gazoles, le procédé d'hydrodésulfuration est primordial afin de respecter les normes environnementales (teneur en soufre inférieure à 10 ppm depuis 2009 en Europe par exemple).
Dans le cas des gazoles, ce procédé est réalisé en réacteur à lit fixe, en régime triphasique (réacteur Trickle-Bed). Les charges réelles contiennent de nombreuses molécules soufrées dont les composés de type thiophénique constituent une classe particulièrement importante [26,27]. Les dibenzothiophènes et les alkyldibenzothiophènes ont été identifiés comme étant parmi les plus réfractaires à l'HDS et sont donc utilisés généralement comme molécules modèles lors des études sur l’HDS de gazoles.
II. 2. 1 - Hydrodésulfuration de molécules soufrées modèles
De nombreuses études [28-36] ont été réalisées sur des molécules modèles telles que le dibenzothiophène (DBT) et le 4,6-diméthyldibenzothiophène (46DMDBT) pour établir les schémas réactionnels et comprendre les mécanismes impliqués en HDS des gazoles.
Ainsi, il a été montré [30,31,36] que ces molécules se transforment selon deux voies : - une voie dite « hydrogénante », notée « HYD », faisant intervenir des étapes d’hydrogénation d’un des deux cycles aromatiques suivie d’une rupture des liaisons C-S selon un mécanisme de type E2
- une voie de désulfuration directe, notée « DSD », conduisant au composé désulfuré par rupture directe des liaisons C-S selon un mécanisme d’élimination E2 (Schéma I-3)
Mo S Mo Mo S Mo S S S S H H H Mo S Mo Mo S Mo S S Mo S Mo Mo S Mo S S S HS S H S
Schéma I-3 : Rupture de la liaison C-S d’un intermédiaire dihydrogéné du dibenzothiophène (voie DSD) [36].
La transformation du DBT (Schéma I-4) conduit majoritairement au biphényle (BPh) selon la voie DSD et au tétrahydrodibenzothiophène (THDBT) et cyclohexylbenzène (CyHBz) selon la voie HYD. A 340°C, sous 3,0 MPa, la sélectivité DSD/HYD mesurée est de l’ordre de 7,0 sur un catalyseur CoMo/Al2O3 [36].
Pour la transformation du 46DMDBT (Schéma I-5), le 3,3’-méthylcyclohexyltoluène (MCyHT) est le produit majoritaire formé selon la voie HYD ainsi que le 4,6-diméthyltétrahydrodibenzothiophène (DMTHDBT). Selon la voie DSD, le seul produit observé est le 3,3’-diméthylbiphényle (DMBPh). A 340°C, sous 3,0 MPa, la sélectivité DSD/HYD mesurée est de l’ordre de 0,2 sur un CoMo/Al2O3 [36].
En outre, récemment des travaux [37,38] ont confirmé ces mécanismes en mesurant la réactivité des différents intermédiaires de la voie HYD. En effet, les intermédiaires tétrahydrogénés et hexahydrogénés (observés pour la transformation du DBT et du 46DMDBT) sont à l’équilibre et la désulfuration peut résulter de leur transformation. De plus, les auteurs ont mis en évidence l’obligation d’une hydrogénation préalable conduisant à un intermédiaire dihydrogéné commun aux voies DSD et HYD.
Ainsi le DBT et le 46DMDBT, qui ont des schémas de transformation similaires, présentent toutefois des contributions de chacune des voies de transformation (HYD et DSD) totalement opposées. La transformation du DBT se fait majoritairement selon la voie DSD et celle du 46DMDBT selon la voie HYD [34,39]. Ceci est expliqué par une gêne stérique due à la présence des groupements méthyles qui inhibe fortement la transformation du 46DMDBT selon la voie DSD [36,40-46].
Le Tableau I-3 rapporte les travaux de différentes équipes, montrant que la présence de groupements méthyles en position adjacente au soufre (c'est-à-dire en positions 4 et 6) est très inhibante. En effet, quel que soit le catalyseur, la température et la pression partielle en H2, le 46DMDBT est beaucoup plus réfractaire à l’HDS que le DBT. En outre, le rapport de
sélectivité DSD/HYD montre bien que la voie DSD est favorisée pour la transformation du DBT et la voie HYD pour la transformation du 46DMDBT.
Tableau I-3 : Comparaison de la transformation du DBT et 46DMDBT en fonction des conditions opératoires.
Auteur Isoda Meille Bataille Egorova
Référence bibliographique 45 42 36 46
Catalyseur NiMo/Al2O3 NiMo/Al2O3 CoMo/Al2O3 CoMo/Al2O3
Température (°C) 320 300 340 340 Pression en H2 (MPa) 2,5 5,0 3,0 5,0 DBT 100 100 100 100 Réactifs 46DMDBT 11 17 17 33 DBT / 6,8 7,0 3,5 Sélectivité DSD/HYD 46DMDBT / 0,1 0,2 0,2
Note : la réactivité du dibenzothiophène a été fixée arbitrairement à 100 pour faciliter la comparaison.
II. 2. 2 - Effet de la pression partielle en sulfure d’hydrogène lors de l'HDS des molécules soufrées réfractaires
L’impact de l’hydrogène sulfuré (H2S) a fait l’objet de nombreux travaux dans la
littérature car il est connu pour être un des principaux inhibiteurs des réactions d’HDS [32]. Il est généralement admis que l’H2S inhibe plus la voie DSD que la voie HYD [2,36,42,46-53].
En effet, il a été observé que les réactions d’hydrogénolyse (conduisant aux ruptures des liaisons C-S) sont plus vulnérables à l’effet inhibiteur de l’H2S que celles d’hydrogénation.
De plus, les catalyseurs à base de molybdène promu par du nickel et par du cobalt ont une affinité vis-à-vis du soufre totalement différente. Il est généralement admis que le NiMo/Al2O3 est plus affecté par la teneur en H2S que le CoMo/Al2O3 [26,46,51,52,54-56].
Contrairement à la tendance générale, Kabe et al. [48] affirment que le NiMo/Al2O3 serait
moins sensible à l’effet inhibiteur de l’H2S que le CoMo/Al2O3. En effet, ils ont comparé
4,6-diméthyldibenzothiophène sur NiMo/Al2O3 et NiW/Al2O3 à d’autres données [57,58]
obtenues sur Mo/Al2O3 et CoMo/Al2O3.
L’impact plus prononcé de l’H2S sur la voie DSD pourrait être dû à des conversions de
sites d’hydrogénolyse (particulièrement utiles à la voie DSD) en sites d’hydrogénation [49,50] ou à une neutralisation préférentielle des sites basiques impliqués dans la voie DSD [36]. L’effet inhibiteur de l’H2S sur les réactions d’HDS et son influence plus marquée sur la voie
DSD pourraient aussi relever de causes mécanistiques et/ou cinétiques, comme le changement d’étapes limitantes [36,59] ou une compétition à l’adsorption [32,46,48,50].
De plus, il est généralement remarqué que l'effet inhibiteur de l’H2S est moins
prononcé lors de l’HDS des alkyldibenzothiophènes que lors de l’HDS du dibenzothiophène [32,36,39,52,60], dû à la plus faible contribution de la voie DSD dans le cas de la désulfuration du 46DMDBT [52]. Enfin, il a été montré [38] que la transformation des intermédiaires tétrahydrogénés et hexahydrogénés du DBT formés par la voie HYD étaient moins sensibles à l’H2S que le DBT lui-même. Le rôle de la pression partielle en H2S dans les
réactions d’HDS est confirmé par des résultats de modélisation cinétique [61]. Par ailleurs, la plus ou moins grande sensibilité des solides NiMo/Al2O3 et CoMo/Al2O3 pourrait provenir de
variations électroniques induites par l’H2S [32,48,54,55]. Des études théoriques semblent
valider cette hypothèse [62].
III - PROCEDES D’HYDRODESOXYGENATION (HDO)
La Commission Européenne impose depuis 2010 que 5,75 % vol. des carburants utilisés à des fins de transport soient des biocarburants [63]. A titre informatif, il est estimé que cet objectif pourra être atteint à condition que 4 à 13 % des surfaces agricoles européennes soient dédiées à la production de biocarburants [64].
Il est donc nécessaire de s’intéresser aux procédés permettant de valoriser cette biomasse en carburant. Un des procédés envisagés pour effectuer cette conversion est la transformation de matière ligno-cellulosique en bio-huiles par pyrolyse, puis la valorisation de ces dernières par un procédé d’HDO permettant de convertir l’oxygène présent dans la coupe majoritairement en H2O, CO et CO2 suivant la nature des fonctions oxygénées présentes
initialement. En effet, cet oxygène provenant de la biomasse se présente sous forme de différentes fonctions chimiques et en proportions élevées (jusqu’à 45 % pds d’oxygène) pour des huiles de pyrolyse [65] (Tableau I-4).
Tableau I-4 : Origine et nature des composés oxygénés.
Origine Fonctions oxygénées Teneur en oxygène
(% pds) Charges pétrolières classiques Phénols Furanes [66] Éthers 0,1 Essences dérivées de la liquéfaction de charbon Phénols Aryl-éthers Benzofuranes (teneurs variées suivant le procédé [67-72]) 4,7 Bio-huiles Méthoxyphénols Cétones Furanes Acides carboxyliques Esters Alcools [65] 16,6 (hydroliquéfiées) 46,9 (pyrolysées)
Pour les bio-huiles, Milne et al. [73], Branca et al. [74], Elliot et al. [75] et Stals et al. [76] ont mis en évidence plus de 400 produits différents, sous forme d'acides, d'alcools, d'aldéhydes, de cétones, d'esters et de composés aromatiques.
III. 1 - Obtention et valorisation de coupes issues de la biomasse ligno-cellulosique Les composés lignocellulosiques peuvent être convertis en carburants liquides par gazéification (production de gaz de synthèse) suivie d’une synthèse Fisher-Tropsch, ou par obtention de bio-huiles ou encore par hydrolyse de la biomasse pour former des sucres sous forme de monomères (Schéma I-6) [77].
La gazéification (obtention de gaz de synthèse) suivi d’un procédé Fisher-Tropsch permet d’obtenir des carburants de grande qualité à partir de ressources hydrocarbonées telles que le charbon ou la biomasse ligno-cellulosique. (Schéma I-7) [78-80]. Si le procédé existe industriellement depuis la seconde guerre mondiale pour le charbon, la fabrication de carburant par cette voie commence seulement à s’implanter à l’échelle du pilote industriel pour la biomasse (SunDiesel en Allemagne). Cependant, le développement de ce type de technique pour la production de biocarburant industriel dans le futur repose essentiellement sur les choix économiques et les stratégies efficaces de conversion de la biomasse en énergie
qui pourront être développées. Le procédé de gazéification permet également la production d’électricité à partir de la biomasse [80].
Schéma I-6 : Stratégies pour la production de carburants à partir de biomasse ligno-cellulosique [77].
Schéma I-7 : Production de carburant liquide et d'électricité à partir de biomasse [80].
Les liquéfiats de biomasse sont obtenus soit par liquéfaction directe à haute pression [81] soit par pyrolyse rapide [82,83](Schéma I-8). La composition de ces huiles dépend du type de procédure utilisée (Tableau I-5).
Biomasse Ligno-cellulosique Gaz de synthèse (CO+H ) Bio-huiles Sucre (aqueux) Lignine EEsssseennccee é étthhéérriiffiiééee É Étthhaannooll H Hyyddrrooccaarrbbuurreessaarroommaattiiqquueess A Allccaanneesslliiqq..oouuhhyyddrrooggèènnee C Caarrbbuurraannttlliiqquuiiddee C Caarrbbuurraannttlliiqquuiiddee Gazéification Pyrolyse ou Liq. Hydrolyse Valorisation Procédé phase aq. Déshydrogénation Zéolite HDO Fermentation H Hyyddrrooggèènnee M Méétthhaannooll A Allccaanneess Fischer-Tropsch Méthanol Water-Gas shift
Schéma I-8 : Système de réacteur de pyrolyse rapide [83].
Tableau I-5 : Propriétés des bio-huiles suivant le procédé utilisé à partir de bois de pin [84].
Procédé utilisé Liquéfaction haute pression Pyrolyse rapide
Teneur en carbone (% pds) 72,6 43,5
Teneur en hydrogène (% pds) 8,0 7,3
Teneur en oxygène (% pds) 16,3 49,2
Densité (g/ml) 1,15 24,8
Pouvoir calorifique (MJ/kg) 35,7 22,6
La liquéfaction directe à haute pression ou procédé HTU® (HydroThermal Upgrading) consiste à faire réagir la biomasse en milieu aqueux à température et pression élevées. La biomasse est successivement traitée dans un premier réacteur à 200°C et 3,0 MPa puis dans un second réacteur à 330°C et 20,0 MPa. En sortie, une phase aqueuse, une phase gaz (H2O et
CO2 principalement) et une huile contenant seulement 10 à 18 % pds d’oxygène sont
obtenues. Ce procédé a l’avantage de pouvoir utiliser une biomasse humide comme charge. Au contraire, pour réaliser une pyrolyse rapide, des étapes de broyage et de séchage sont généralement nécessaires avant le procédé de liquéfaction à proprement parler pour obtenir une biomasse fine (granulométrie < 6 mm) et sèche (humidité < 10 %). Le procédé requiert ensuite une chauffe rapide (100 à 500 °C/s), une température modérée (500 °C), un temps de séjour des vapeurs court (< 2 s) et une pression faible (< 1 bar) pour maximiser le rendement en liquide. Cependant, ces huiles possèdent une teneur très élevée en composés oxygénés (40 à 50% massique), les rendant inutilisables en l’état. En effet, cette forte teneur
en oxygène leurs confère une mauvaise stabilité au cours du temps due à un accroissement de leur viscosité [65,85]. De plus, leur pouvoir calorifique est environ deux fois plus faible que celui d’un fuel (15 à 20 MJ/kg pour une huile contre 40 MJ/kg pour un fuel). Elles possèdent enfin un pouvoir corrosif élevé. Il est par conséquent nécessaire de leur faire subir un traitement adapté avant de pouvoir les commercialiser comme carburants ou comme combustibles.
Il est ainsi possible de les valoriser par un procédé d’hydrodésoxygénation avant de les utiliser comme carburant. Cette valorisation peut se faire soit par un procédé d’HDO de l’huile brute, soit par un co-traitement avec une coupe gazole ce qui permettrait de réaliser simultanément l’hydrodésulfuration de la coupe gazole et l’hydrodésoxygénation de l’huile de pyrolyse et d’utiliser les unités d’hydrotraitement actuellement en service dans les raffineries.
Tableau I-6 : Composition d’une huile de pyrolyse rapide par rapport à un brut pétrolier classique [65].
Composition d’un pétrole brut (% pds)
Composition d’une huile de pyrolyse (% pds) Carbone 85,2 45,3 Hydrogène 12,8 7,5 Soufre 1,8 < 0,1 Azote 0,1 < 0,1 Oxygène 0,1 46,9
III. 2 - Procédé d’hydrodésoxygénation des huiles brutes
Le procédé d'HDO vise ainsi à éliminer l'oxygène présent dans les liquéfiats sous forme d’H2O, de CO et/ou de CO2, suivant la nature des fonctions oxygénées présentes
initialement. Les conditions opératoires sont généralement comprises entre 300 et 600°C, sous une pression élevée en H2 [65]. Ce procédé d’HDO peut s'effectuer en présence de catalyseurs
sulfurés CoMo/Al2O3 ou NiMo/Al2O3, mais aussi en présence de catalyseurs à base de
Pt/SiO2 [86], VN [87] et Ru [88]. Les bio-huiles valorisées peuvent atteindre un indice
d’octane Recherche (RON) de 77 [65].
Elliot et al. [84,89-91] ont développé un procédé d’hydrotraitement en 2 étapes pour la valorisation des bio-huiles dérivées de pyrolyse de bois de pin. La première étape est un traitement catalytique (270°C, 13,6 MPa) qui va permettre l’hydrogénation des composés
instables (non précisés) présents dans les bio-huiles pour les stabiliser. La seconde étape consiste en une hydrogénation catalytique à haute température (400°C, 13,6 MPa). Le même catalyseur (CoMo/Al2O3 sulfuré ou NiMo/Al2O3 sulfuré) est utilisé pour les 2 étapes. Ce
procédé permet d’atteindre des rendements massiques de 40 % avec une huile raffinée contenant moins de 1 % pds d’oxygène. Durant ce procédé, 20-30 % pds du carbone présent dans les bio-huiles est converti en CO et CO2, ce qui est à l’origine du faible rendement
obtenu.
Su-Ping et al. [92] ont étudié l’hydrogénation d’une huile (dont la provenance n’est pas précisée) contenant 32 % pds d’oxygène. La réaction est effectuée dans un autoclave à des pressions partielles d’hydrogène comprises entre 1,5 et 3,0 MPa et à des températures comprises entre 360 et 390 °C, en présence d’un catalyseur CoMoP/Al2O3. Après 30 min de
réaction, la désoxygénation est maximale et atteint environ 90 % dans tous les cas. Cette désoxygénation augmente légèrement avec la température (5 % de plus à 390°C qu’à 360°C). La pression partielle en hydrogène, dans les conditions étudiées, n’affecte pas la désoxygénation. De même, de Miguel Mercader et al. [93] ont étudié l’hydrotraitement d’une huile de pyrolyse issue de bois de pin dans un réacteur tubulaire entre 200 et 350°C et entre 20 et 25 MPa. Le pouvoir calorifique de l’huile augmente de 17 à 28 MJ/kg après hydrotraitement avec des rendements en huile hydrotraitée de l’ordre de 60 % pds, le complément étant principalement une phase aqueuse.
Cependant ces réactions sur des huiles brutes restent difficiles en raison de leur grande instabilité due à la forte teneur en oxygène. De plus, leur haute viscosité nécessite un préchauffage et une mise sous pression préalable pour les fluidiser et ainsi pouvoir les utiliser. Par conséquent, la technologie actuellement développée pour l’hydrotraitement des coupes pétrolières ne semble pas adaptée au traitement d’huiles de pyrolyse de biomasse [94].
III. 3 - Transformation de charges modèles de bio-huiles
De nombreux travaux ont été réalisés dans les années 90 et ces dernières années sur l’HDO de molécules oxygénées modèles.
Delmon et al. [95-100] ont étudié l’hydrodésoxygénation de composés modèles d’une bio-huile (4-méthylacétophénone, décanoate d’éthyle et guaiacol) sur catalyseurs CoMo et NiMo sulfurés supportés sur alumine ou sur support carboné, de manière à déterminer les mécanismes réactionnels, l’influence des paramètres principaux de réaction (pression totale,
température, temps de réaction, …), et enfin les possibles poisons pour ces catalyseurs (Schéma I-9).
La fonction cétone est facilement et sélectivement hydrogénée en une fonction méthylène à 200°C [95]. Les fonctions acides carboxyliques sont elles aussi hydrogénées dans les conditions d’HDO, mais une réaction parallèle de décarboxylation se produit aussi à des taux comparables [95]. Les acides carboxyliques ainsi que le guaiacol et ses dérivés ne sont cependant pas aussi réactifs que les groupements cétones, et des températures supérieures à 300°C se révèlent nécessaires pour leurs conversions. Le guaiacol est hydrogéné en catéchol, puis en phénol, mais il conduit par ailleurs à une désactivation du catalyseur par réaction de cokage (12 % pds de carbone).
Schéma I-9 : HDO de charges modèles de bio-huiles [97].
L’acidité du support ne change pas le taux d’hydrogénation de la 4-méthylacétophénone, mais augmente les taux de décarboxylation et d’hydrogénation du décanoate d’éthyle et la formation de coke à partir du guaiacol [97]. Le carbone, qui présente une faible acidité, se révèle être un support adapté pour les réactions d’hydrodésoxygénation [97].
III. 4 - Transformation de différentes molécules modèles
III. 4. 1 - Le benzofurane
La transformation du benzofurane a fait l’objet de nombreux travaux. Satterfield et Yang [101] ont réalisé la transformation du benzofurane à 375°C sous 6,9 MPa sur un NiMo/Al2O3 et en présence de CS2 comme agent sulfurant. Ils ont observé de
l’éthylcyclohexane et de l’éthylméthylcyclopentane comme principaux produits de désoxygénation du benzofurane (de l’éthylbenzène est aussi détecté mais en faibles quantités). Bunch et Ozkan [102] ont quant à eux étudié la décomposition du benzofurane à différentes températures (200-360°C) et pressions (2,0 et 5,5 MPa), dans un réacteur à lit fixe en présence de NiMo/Al2O3 commercial. A 320°C, sous 5,5 MPa, une conversion de 93,4 % a
été obtenu avec majoritairement de l’éthylcyclohexane (52,5 %), du cyclohexane (22,3 %) et de l’éthylphénol (17,1 %). La formation du méthylcyclohexane (5,3 %), du dihydrobenzofurane (1,1 %), de l’éthylbenzène (0,9 %) et de l’éthylcyclohexène (0,5 %) a également été observée.
L’influence de la nature du catalyseur sur l’HDO du benzofurane à 370°C sous 3,1 MPa, en présence de NiMo/Al2O3 commercial ou de nouveaux catalyseurs (Mo2C, WC,
Mo2N, VN, NbC, VC et TiN) a aussi été mise en évidence par Ramanathan et Oyama [87]. Le
catalyseur NiMo/Al2O3 serait le plus actif, les composés majoritaires formés étant dans
chaque cas l’éthylbenzène et l’éthylcyclohexane.
Une conversion quasi-totale du benzofurane est observée avec, comme principaux produits, le benzène, le toluène et l’éthylbenzène sur Mo2N à 400°C sous une pression en H2
proche de la pression atmosphérique [103].
Plus récemment, Romero et al. [104] ont établi le schéma de transformation du benzofurane dans les conditions classiques d’hydrotraitement (à 340°C sous 7,0 MPa sur un NiMoP/Al2O3) (Schéma I-10). Tout d’abord le benzofurane est rapidement hydrogéné en
dihydrobenzofurane qui, par rupture de la liaison C-O, conduit à la formation du 2-éthylphénol. Celui-ci peut se désoxygéner selon 3 voies :
1/ une voie dite « hydrogénante » (HYD), voie majoritaire (71 % de sélectivité), conduisant à l’éthylcyclohexane, produit majoritaire de réaction, l’intermédiaire éthylcyclohexanol n’étant pas observé,
2/ une voie dite de « désoxygénation directe » (DDO), voie la plus minoritaire (7 % de sélectivité), conduisant à l’éthylbenzène par rupture directe de la liaison C-O,
3/ une voie de dismutation et d’isomérisation (ACI), voie minoritaire (23 % de sélectivité), conduisant au phénol et aux diéthylphénols par dismutation et au 3-éthylphénol par isomérisation, des composés désoxygénés de ces produits étant également observés mais en faibles quantités.
Enfin, ces auteurs ont montré que la faible réactivité du benzofurane serait due à une inhibition de la transformation du 2-éthylphénol par la présence de benzofurane lui-même ou de son intermédiaire hydrogéné, le 2,3-dihydrobenzofurane.
Schéma I-10 : Transformation du benzofurane sur NiMoP/Al2O3 [104].
III. 4. 2 - L’heptanoate de méthyle
Senol et al. [105-109] ont étudié la décomposition de l’heptanoate de méthyle à 250°C, sous 1,5 MPa, en présence de NiMo/Al2O3 et CoMo/Al2O3 sulfurés. Il a alors été
montré que sur NiMo/Al2O3, la formation d'hydrocarbures saturés était majoritaire alors que
sur CoMo /Al2O3, il y a principalement formation d'hydrocarbures insaturés. Le NiMo/Al2O3
conduit à la formation d'heptane comme principale produit avec une sélectivité de 43%, tandis que le CoMo/Al2O3 conduit essentiellement à la formation de 35% d'heptènes (Figure I-9).
Figure I-9 : Distribution des produits formés par décomposition de l’heptanoate de méthyle sur catalyseurs sulfures [108].
Considérant les produits formés au cours de la décomposition de l’heptanoate de méthyle, le schéma réactionnel suivant a pu être établi (Schéma I-11) où apparaissent deux voies différentes:
- une voie d'hydrolyse de l'ester en acide, qui peut être suivie soit par une étape d'estérification, soit par des étapes successives de déshydratation/décarboxylation conduisant à un alcane à 6 atomes de carbone,
- une voie d'hydrogénation conduisant à un alcool qui par déshydratation forme une oléfine qui peut ensuite s’hydrogéner en alcane à 7 atomes de carbone.
La transformation de l’acide heptanoïque, principal intermédiaire de la transformation de l’ester [109] a également été effectuée (1,5 MPa, 250°C en présence d’H2S).
Schéma I-11 : Transformation de l’heptanoate de méthyle sur catalyseur NiMo/Al2O3 et
Les produits observés sont soit l’heptane et les heptènes (« C7 ») obtenus par décarboxylation ou décarbonylation, soit l’hexane et les hexènes (« C6 ») obtenus par hydrogénation puis déshydratation. Un rapport molaire C7/C6 de 2,0 et un rapport alcane/alcène de 0,2 pour le CoMo/Al2O3 d’un part et un rapport molaire C7/C6 de 1,6 et un
rapport alcane/alcène de 4,2 pour un catalyseur NiMo/Al2O3 d’autre part ont été chiffrés. La
décarbonylation ou la décarboxylation sont minoritaires par rapport à l’hydrogénation de l’acide. Ils ont également observé de très faibles quantités d’heptanal, d’heptanol, de diheptyléther, d’heptanoate d’heptyle, d’heptan-1-thiol et de hexan-1-thiol.
III. 4. 3 - Les composés phénoliques
La transformation du phénol et de ses dérivés a fait l’objet de nombreux travaux dans la littérature [110-118]. En effet, ces réactions de désoxygénation feraient intervenir deux voies de transformation :
- une voie de rupture directe de la liaison carbone-oxygène (voie DOD) conduisant à une élimination directe de l'atome d'oxygène par perte d’eau,
- une voie nécessitant l’hydrogénation du noyau aromatique (voie HYD) conduisant à un intermédiaire de type cyclohexanol, suivie de l'élimination de l'oxygène sous forme d'eau (voie HYD) (Schéma I-12).
OH
OH
HYD DOD
Schéma I-12 : Hydrotraitement du phénol [110].
L'intermédiaire cyclohexanol se convertit très vite et n'est généralement pas observé. La sélectivité DOD/HYD dépend des conditions opératoires et du catalyseur étudié. En effet, des sélectivités DOD/HYD pour la transformation du phénol de 13 et 6 sont respectivement observées à 250 et 300°C pour une pression totale de 1,5 MPa sur un CoMo/Al2O3 [110]. En
revanche, dans des conditions plus proches de celles de l’HDS du 4,6-diméthyldibenzothiophène à 7,8 MPa et 225°C sur un CoMo/Al2O3, ce rapport est de 0,6
La présence de groupes méthyles en ortho induisent une gêne stérique qui inhibe les réactions d'hydrodésoxygénation [111,113,120]. Par ailleurs, le m-crésol est décrit comme ayant une réactivité proche de celle du phénol alors que la présence du groupement méthyle en position para induit une stabilisation du groupement phénolique le rendant ainsi moins réactif. Les réactivités relatives de l'o-crésol, du m-crésol et du p-crésol ont été mesurées par Massoth et al. [111]. Le p-crésol est l'isomère le plus réactif tandis que l’o-crésol présente la réactivité la plus faible. En revanche, Odedunmi et Ollis [120] ont observé à 350°C sous 6,8 MPa et en présence d’un CoMo/Al2O3 que le m-crésol était plus réactif que le p-crésol,
l'o-crésol étant également l’isomère le moins réactif.
Romero et al. [119] ont étudié la transformation du 2-éthylphénol sur catalyseur Mo/Al2O3, NiMo/Al2O3 et CoMo/Al2O3 supportés sur alumine (7,0 MPa, 340°C). La
promotion du catalyseur par le cobalt favorise la voie DOD alors que la promotion par le nickel favorise la voie HYD. Il a également été mis en évidence que la déshydratation de l’alcool, intermédiaire de la voie HYD, avait lieu sur l’alumine.
Le guaiacol (2-méthoxyphénol) est un des composés oxygénés majoritaires des huiles de pyrolyse de biomasse et a été étudié notamment par Kallury et al. [114] et Delmon et al. [95-97,99,115]. Les expériences ont été réalisées dans un autoclave à 250°C sous 7,0 MPa. Sur alumine seule, le catéchol est le seul produit formé [96]. Ces auteurs ont alors proposé comme hypothèse que les sites acides de Lewis de l'alumine permettent la déméthylation du guaiacol, ce qui est corroboré par l'absence d'inhibition de cette première étape en présence d'eau ou d'H2S et par le fort effet inhibiteur de l'ammoniac [96]. En présence des catalyseurs
CoMo/Al2O3 et NiMo/Al2O3, la formation de catéchol et de phénol est observée dans un
premier temps, le phénol étant ensuite transformé en benzène et en cyclohexane (Schéma I-13) [96]. Le bilan massique d'environ 80% observé, obtenu lors de l'hydrotraitement du guaiacol avec un catalyseur CoMo/Al2O3, est expliqué par la formation de produits lourds et
de coke à partir du guaiacol ou du catéchol.
OH OMe
OH OH
OH
GUA CAT PhOH
coke
Bz
CyH
L'hydrotraitement du guaiacol en autoclave sur CoMo/Al2O3 à 250°C sous 3,45 MPa a
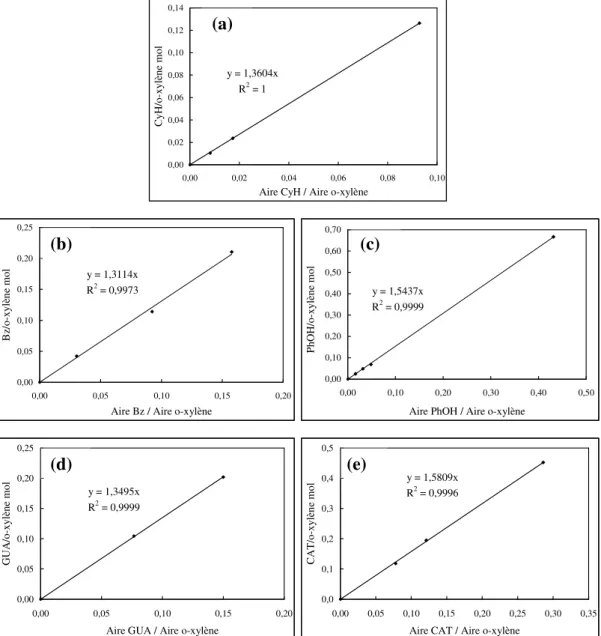
montré que le catéchol, produit primaire de la réaction, se transforme rapidement en phénol qui conduit ensuite au cyclohexane et benzène avec une sélectivité cyclohexane/benzène d'environ 2 (Figure I-10) [116]. Ces résultats sont en accord avec le schéma proposé par Delmon et al. [96].
Figure I-10 : Rendement des principaux produits en fonction du temps lors de l'hydrotraitement du guaiacol à 250°C sous 3,45 MPa [116].
De plus, il peut y avoir un effet électronique positif du groupe hydroxyle en position ortho sur la réaction de déméthylation. En effet, celle du guaiacol est 30 fois plus rapide que celle de l'anisole (méthoxybenzène) dans les mêmes conditions [116].
L'hydrotraitement poussé du guaiacol conduit aux mêmes produits que ceux observés lors de la transformation du catéchol. Des produits de méthylation (o-crésol, toluène, méthylcyclohexane et méthylcatéchol) sont aussi observés en faibles quantités, attribués à la présence de méthane issu de la première déméthylation. Kallury et al. [114] observent également la présence de toluène et de xylènes.
Ferrari et al. [97] ont étudié l'hydrotraitement du guaiacol dans un réacteur à lit fixe contenant du catalyseur CoMo supporté sur du carbone activé. Avec un débit de charge de 45 mL/h, une température de 270°C et une pression de 7,0 MPa, ils obtiennent une conversion du guaiacol de l'ordre de 20 % mol avec environ 5 % mol de catéchol. Aucune influence de la pression partielle en H2S sur la transformation du guaiacol n’a été mise en évidence.
Cependant, ils ont pu remarquer que l’H2S avait un effet inhibiteur sur la conversion du
catéchol en phénol. De plus, il a pu être montré qu'en présence de catalyseur NiMo supporté sur carbone, la conversion était bien plus faible que lorsque les métaux sont supportés sur
alumine. Ceci confirme l'hypothèse d'une déméthylation catalysée en partie par les sites acides de Lewis de l'alumine [97,117].
Vuori et al. [118] ont proposé des schémas d'adsorption du guaiacol sur des catalyseurs sulfures (Schéma I-14) et de formation du catéchol et du phénol (Schéma I-15) en présence de CoMo/Al2O3 dans un réacteur à lit fluidisé.
S S M S S CH3 M OH O CH3 OH O
Schéma I-14 : Adsorption du guaiacol sur des catalyseurs sulfures [118].
S H M OH O S M OH OH S S S H M OH O S M OH S S O A. B.
Schéma I-15 : Formation du catéchol (A) et du phénol (B) [118].
III. 4. 4 - Transformation du CO – Réaction de water gas shift
De nombreux travaux sont consacrés à la réaction de water gas shift, néanmoins cette réaction a été peu étudiée sur des catalyseurs d’hydrotraitement.
La réaction de water gas shift (Equation 1) est depuis très longtemps utilisée afin de convertir le CO présent dans les gaz de reformage en hydrogène [121].
2 2
2O CO H
H
Cette réaction présente plusieurs avantages lorsqu’elle est utilisée dans un système catalytique couplé à une unité de reformage. En effet, on obtient de bonnes activités à faible température (inférieure à 250°C), une stabilité vis-à-vis de différents poisons (H2S, chlore),
une bonne stabilité dans les conditions de reformage et les réactions secondaires telles que la méthanation ne sont pas contraignantes [121].
La réaction de water gas shift est exothermique et réversible. Comme sa constante d’équilibre diminue avec l’augmentation de la température, il est préférable de se placer à de basses températures pour augmenter la conversion de CO. Les catalyseurs les plus couramment utilisés sont des Fe2O3-Cr2O3 pour des transformations à hautes températures
(350-420°C) et des Cu-ZnO/Al2O3 pour des transformations à basses températures
(180-270°C) [122,123]. Comme une vapeur de reformage contient en moyenne 10 % mol. de CO, le procédé à haute température permet d’en convertir environ 60 %. Ensuite, le procédé à basse température permet d’abaisser la teneur en CO à 0,4 % mol en profitant d’un équilibre thermodynamique plus favorable. Ces deux procédés sont donc complémentaires. Différentes études cinétiques [124,125] ont permis de proposer deux mécanismes réactionnels, un mécanisme régénératif et un mécanisme par adsorption (Figure I-11).
(a) (b)
Figure I-11 : Mécanisme régénératif (a) et mécanisme par adsorption (b) pour la réaction de water gas shift.
Beaucoup d’autres catalyseurs, notamment Mo2C [126,127], des oxydes de Mo, V et
W [128] ou encore des métaux nobles supportés sur CeO2 [129,130], ont été envisagés. Le
Mo2C est connu pour être sensible à l’oxygène et son activité est pour l’instant peu étudiée.
Les oxydes de Mo, V et W sont utilisés car ils ne catalysent pas la réaction parasite de méthanation. Les catalyseurs à base de métaux nobles présentent une grande activité sur une large gamme de température mais ne sont pas économiquement viables.
Le sulfure de molybdène a également été étudié pour sa tolérance à l’H2S [131]. En
classiques. Les CoMo/Al2O3 et NiMo/Al2O3 sulfurés présentent une activité pour la réaction
de water gas shift intéressante à haute température (450°C) (40 % de conversion de CO pour une VVH de 8 000 h-1) mais nécessitent un système complémentaire pour être totalement efficaces industriellement afin d’éliminer le CO restant. Ces catalyseurs sont toutefois utiles pour convertir une première partie du CO si de l’H2S est présent dans le mélange [132].
III. 5 - Influence des sous-produits de réaction sur l’HDO de molécules oxygénées Selon les molécules oxygénées à hydrotraiter (fonctions acides, cétone, aldéhyde, ester, alcool) ou les conditions opératoires (présence d’H2S), les produits secondaires formés
sont différents. En effet, l’hydrodésoxygénation d’alcools aliphatiques ou aromatiques conduit à la formation d’eau comme produit secondaire alors que la présence d’H2O, de CO
et/ou de CO2 est observée à partir des fonctions acide ou ester. Peu d’études relatent dans la
littérature l’effet de la présence de ces sous-produits sur la modification des propriétés catalytiques des catalyseurs.
Des conclusions contradictoires dans la littérature ont été établies sur les effets que pouvait présenter l’eau vis-à-vis des réactions d’HDO. Ainsi, lors de la transformation du dibenzofurane, sur un catalyseur NiMo/Al2O3 sous une pression totale de 6,9 MPa, La Vopa
et Satterfield [133] ont montré qu’une pression partielle en eau de 24 kPa n’affectait pas le taux d’HDO en présence de l’H2S, et accélérait même la réaction en absence d’H2S. En
revanche, Vogelzang et al. [134] ont observé une inhibition de l’HDO du 1-naphtol sur catalyseurs Mo/Al2O3 et NiMo/Al2O3 par l’eau formée au cours de la réaction. Des études
plus récentes réalisées par Laurent et al. [96,135] rapportent que l’addition d’eau provoque seulement une faible inhibition de l’HDO des cétones, des acides carboxyliques et du phénol sur catalyseurs NiMo/Al2O3 et CoMo/Al2O3. Par ailleurs, Laurent et al. [100] observent
qu'une pression partielle de 2,5 MPa en eau est responsable de la perte des deux tiers de l’activité initiale d'un catalyseur NiMo/Al2O3 en 60h au cours de l’HDO d'un mélange de
4-méthylphénol et de dibenzofurane sous 7 MPa de pression totale.
Senol et al. [105] ont examiné l’influence de l’eau sur la transformation de l’heptanoate d’éthyle. L'eau est un inhibiteur des réactions de désoxygénation (diminution de la conversion des esters de près de 10 % pour un catalyseur CoMo/Al2O3 et de près de 15%
pour un catalyseur NiMo/Al2O3 avec 18 600 ppm d'eau) et plus particulièrement la réaction de
décarboxylation (impossibilité de former les C6-hydrocarbures). Il semble que l’eau affecte
NiMo/Al2O3 mais pas de changement notable avec le CoMo/Al2O3. Il est alors intéressant
d'ajouter un agent sulfurant comme l’H2S qui va préserver l'activité catalytique en
compensant l'effet inhibiteur d’H2O. La quantité d’H2S devra néanmoins être rationalisée,
Senol et al. ayant utilisé un mélange contenant 830 ppm d'H2S et 1,86 % pds d’H2O.
Les travaux de la littérature rapportant l'influence de l’H2S et d’agents sulfurants au
sens général sur les réactions d'HDO conduisent aussi à des conclusions très contradictoires. Senol et al. [108] précisent tout d’abord que les effets de l'addition d'un agent sulfurant à une charge à désoxygéner afin de maintenir la stabilité et l’activité du catalyseur utilisé dépendent grandement de la composition de la bio-huile de départ. En effet, l’addition d’un agent sulfurant à une charge à base de bois, qui ne contiendra principalement que des composés phénoliques, aura un effet négatif en termes d’HDO. A l’opposé, un effet promoteur est notable dans le cas du traitement d’huiles végétales ou de graisses animales qui contiennent principalement des composés oxygénés aliphatiques. Ainsi, dans une autre étude, Senol et al. [107] précisent l’effet promoteur d’H2S sur l'activité de catalyseurs sulfurés
NiMo/Al2O3 et CoMo/Al2O3 pour la conversion d'esters aliphatiques (250°C, 15 MPa). Ces
auteurs observent un gain de l’ordre de 10 % de conversion avec un catalyseur NiMo/Al2O3 et
de 20 % de conversion avec un catalyseur CoMo/Al2O3, mais sans pour autant prévenir toute
désactivation. L’addition d’H2S favorise les réactions catalysées en milieu acide en
augmentant l’acidité du catalyseur. Par ailleurs, l’H2S inhibe les réactions d’hydrogénation
sur le catalyseur NiMo/Al2O3, mais n’influe pas sur l’activité du catalyseur CoMo/Al2O3.
L’addition d'H2S entraîne la formation de C6-hydrocarbonés, et augmente par la même
occasion la formation d’oxydes de carbone (CO et CO2). A noter que la concentration en H2S
est un paramètre important à prendre en compte, car elle va influer sur l’HDO globale, sur la consommation en hydrogène et enfin sur les degrés de saturation des molécules formées.
Des travaux de Bunch et al. [136] relatent l’influence de l’H2S sur l’HDO du
benzofurane. Ils ont conclu que la présence d’H2S dans la charge conduisait à une diminution
de l’HDO du benzofurane (de l’ordre de 10 à 20 % suivant les conditions opératoires) pour un catalyseur NiMo/Al2O3 sulfuré ou réduit, ceci résultant principalement de l'adsorption
compétitive entre l’H2S et les espèces benzofuraniques à la surface du catalyseur.
Enfin, Romero et al. [104] ont observé un effet promoteur d’H2S sur les réactions de
désoxygénation du benzofurane, du dihydrobenzofurane et du 2-éthylphénol (à 340°C sous 7 MPa, NiMoP/Al2O3). En revanche, l’H2S inhibe la formation d’éthylbenzène (voie DOD) à

![Tableau I-5 : Propriétés des bio-huiles suivant le procédé utilisé à partir de bois de pin [84]](https://thumb-eu.123doks.com/thumbv2/123doknet/7962523.266782/30.892.263.630.80.364/tableau-propriétés-huiles-suivant-procédé-utilisé-partir-bois.webp)