ÉTUDE DE LA FOCALISATION D’UN FAISCEAU IONIQUE À LA
PRESSION ATMOSPHÉRIQUE PAR DES LENTILLES
ÉLECTROSTATIQUES ANNULAIRES MINCES, DANS UNE
DÉCHARGE COURONNE EN CONFIGURATION POINTE-PLAN
Mémoire
Jonathan Rochon
Maîtrise en Physique
Maître ès sciences (M.Sc.)
Québec, Canada
© Jonathan Rochon, 2013
iii
Résumé
Ce projet vise l’étude de la focalisation d’un faisceau ionique à la pression atmosphérique. Les sources ioniques récentes fonctionnent à cette pression et la région de dérive séparant la région d’ionisation de l’entrée du spectromètre de masse est telle qu’une importante partie du signal produit est perdu avant son introduction dans le spectromètre de masse. La source LDTD ionise les analytes par une décharge couronne qui est produite par une pointe à haute tension. Le champ électrique généré par cette configuration tend à grandement disperser les ions. Pour limiter cette dispersion et focaliser une plus grande partie du signal produit au centre, des lentilles électrostatiques annulaires minces ont été introduites dans la région de dérive. Des simulations ainsi que des expériences réalisées sur un montage, nous ont permis d’observer une focalisation d’un faisceau ionique. Les résultats expérimentaux ont corroboré les résultats obtenus en simulation.
v
Table des matières
RÉSUMÉ ... III TABLE DES MATIÈRES ... V LISTE DES FIGURES ... IX LISTE DES TABLEAUX ... XIII LISTE DES SYMBOLES MATHÉMATIQUES ...XV REMERCIEMENTS ... XIX CHAPITRE 1 : INTRODUCTION ... 11.1. HISTORIQUE DE LA SPECTROMÉTRIE DE MASSE ET BUT DU PROJET ... 1
1.2. OBJECTIFS DU PROJET ET MÉTHODOLOGIE ... 7
CHAPITRE 2 : REVUE DES TECHNOLOGIES ET CONCEPTS ... 11
2.1 INTRODUCTION ... 11
2.2 LES MÉTHODES D’IONISATION UTILISÉES DANS LE VIDE... 13
2.2.1 L’IONISATION PAR BOMBARDEMENT ÉLECTRONIQUE ... 14
2.2.2 L’IONISATION CHIMIQUE ... 16
2.2.3 L’IONISATION DE SURFACE ... 18
2.2.4 L’IONISATION PAR EFFET DE CHAMP ... 19
2.2.5 L’IONISATION PAR PLASMA ... 20
2.2.6. RÉSUMÉ ... 21
2.3 LES SOURCES ET MÉTHODES D’IONISATION À LA PRESSION ATMOSPHÉRIQUE ... 22
2.3.1 L’ÉLECTRO-NÉBULISATION ... 24
2.3.3 LA SOURCE LDTD ... 28
2.3.4 RÉSUMÉ ... 30
2.4 LA DÉCHARGE COURONNE, LENTILLES ÉLECTROSTATIQUES ET TRANSPORT IONIQUE À LA PRESSION ATMOSPHÉRIQUE ... 31
2.4.1 CARACTÉRISATION DE LA DÉCHARGE COURONNE ... 32
2.4.2 LENTILLES ÉLECTROSTATIQUES ... 41
2.4.3 MOBILITÉ IONIQUE ET DIFFUSION... 43
CHAPITRE 3 : SIMULATION NUMÉRIQUE ... 49
3.1 MÉTHODES NUMÉRIQUES POUR CALCULER LE POTENTIEL ET LE CHAMP ÉLECTRIQUE... 51
3.2 RELAX 3D ... 55
3.2 SIMION ET SDS ... 57
3.3 SIMULATIONS ET RÉSULTATS ... 60
3.3.1 LIMITES ... 61
3.3.2. COMPARAISON DU COMPORTEMENT IONIQUE SOUS DIVERSES VALEURS DE PRESSION ... 65
3.3.3 ÉTUDE DE LA CONFIGURATION POINTE-PLAN AVEC DIVERSES LENTILLES ÉLECTROSTATIQUES : RÉSULTATS ET ANALYSES ... 68
CHAPITRE 4 : MONTAGE EXPÉRIMENTAL ... 83
4.1 ASSEMBLAGE DES COMPOSANTES DU MONTAGE ... 83
4.2 ISOLATION OPTIQUE ENTRE LES DÉTECTEURS ET L’ORDINATEUR ... 88
4.2.1 CIRCUIT DU MODULE ÉMETTEUR (ÉLECTROMÈTRE) ... 91
4.2.2 CIRCUIT DU MODULE RÉCEPTEUR ... 95
4.2.3 CIRCUIT DE L’AMPÈREMÈTRE ... 96
4.2.4. RÉSUMÉ ... 97
4.3 CONTRÔLE DU MONTAGE PAR LABVIEW ... 98
4.4 CONCLUSION... 99
CHAPITRE 5 : RÉSULTATS EXPÉRIMENTAUX ET DISCUSSIONS ... 101
vii
5.2 VALIDATION DU MONTAGE ET PREMIÈRES MESURES ... 105
5.3 RÉSULTATS PRÉLIMINAIRES RECUEILLIS MANUELLEMENT ... 108
5.4 EXPÉRIMENTATION DE LENTILLE SIMPLE DE TYPE ‘’EV PARTS’’ : RÉSULTATS ET DISCUSSIONS. ... 113
5.4.1. EFFET DU POSITIONNEMENT DES LENTILLES DE 19,1 MM ET 9,6 MM SUR LA FOCALISATION DES IONS. .. 115
5.4.2. DIMINUTION DU COURANT IONIQUE DÉTECTABLE SUR LES DÉTECTEURS PRODUITE PAR LA LENTILLE ÉLECTROSTATIQUE. ... 120
5.4.3. COMPARAISON DE LA FOCALISATION ENTRE LES DIVERSES LENTILLES UTILISÉES. ... 126
5.4.4 RÉSUMÉ ... 133 5.5 QUELQUES RÉFLEXIONS ... 135 5.5.1. LENTILLES MULTIPLES ... 135 5.5.2. GAZ VECTEUR ... 137 CONCLUSION ... 137 RÉFÉRENCES ... 143 ANNEXE A ... 147 ANNEXE B ... 149 ANNEXE C ... 151 ANNEXE D ... 153
ix
Liste des figures
Figure 1.1: Densité relative du courant ionique en fonction de l'angle (rad) ... 8Figure 2.1: Efficacité d'ionisation selon l'énergie des électrons 71 ... 15
Figure 2.2: Interface à la pression atmosphérique 16 ... 23
Figure 2.3: Principe de fonctionnement d’une source à électro-nébulisation 17 ... 25
Figure 2.4: Schéma de fonctionnement d’une source MALDI 73. ... 27
Figure 2.5: Schéma de fonctionnement de la source LDTD ... 28
Figure 2.6: Effet de charge aux alentours de la pointe pour la décharge couronne négative 20 ... 34
Figure 2.7: Courant produit par la décharge couronne en fonction de la tension appliquée à la pointe pour un espacement de 2 cm 20 ... 35
Figure 2.8: Courant minimum détectable avec la tension correspondante en fonction de l’espacement d 21 ... 36
Figure 2.9: Courant produit par la décharge couronne en fonction du voltage appliqué à la pointe pour différentes valeurs de d 21 ... 37
Figure 2.10: Système de lentilles électrostatiques en forme d’entonnoir utilisé en spectrométrie par mobilité ionique 32 ... 42
Figure 2.11: Schéma de montage avec les équipotentielles (a) sans lentille et (b) avec une lentille électrostatique positionnée derrière la pointe d’électro-nébulisation 33. ... 42
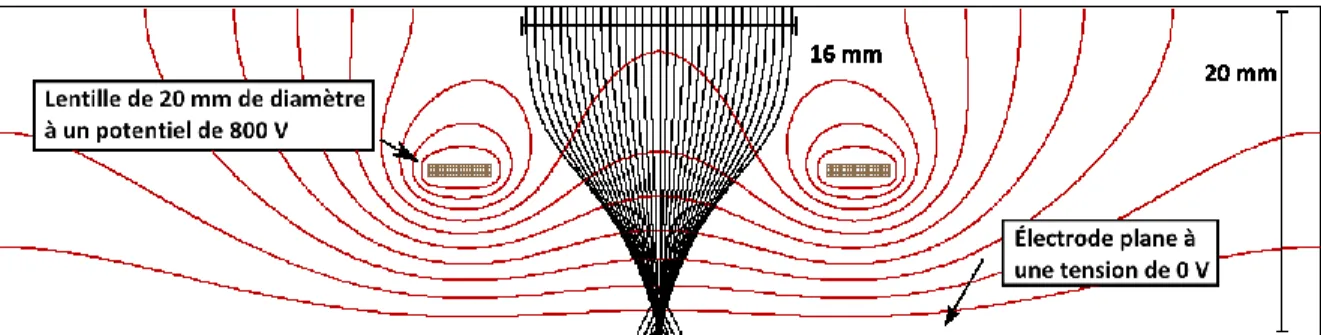
Figure 2.12: Lentille électrostatique de forme ellipsoïdale utilisée avec l’électro-nébulisation 34 ... 43
Figure 3.1: Nœuds adjacents au nœud central en x, y et z. ... 52
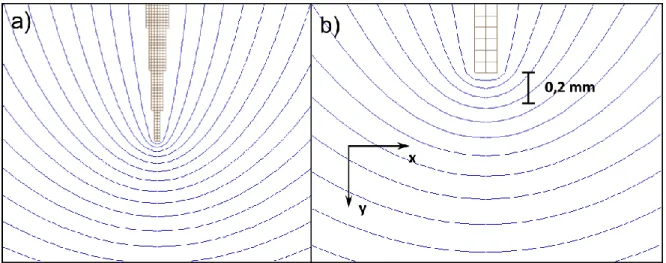
Figure 3.2: a) Schéma d’une électrode rectangulaire ayant 1 mm de large et 6 mm de haut face à une seconde électrode plane. b) Agrandissement sur la pointe de cette électrode (environ 5mm de haut sur 7 mm de large). ... 62
Figure 3.3: a) Les deux premiers cercles sont dessinés dans un espace de 100 nœuds, le premier ayant un rayon de 1 nœuds et le second un rayon de 3 nœuds. b) Le troisième a été réalisé dans un espace de 1000 nœuds et a un rayon de 29 nœuds. ... 63
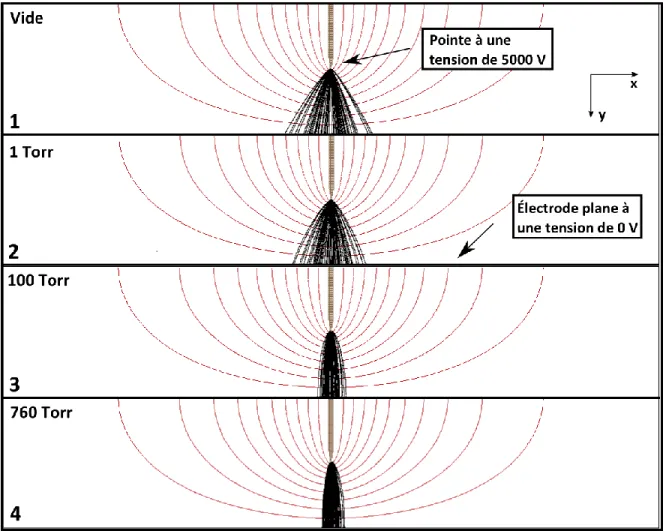
Figure 3.4: Pointe ayant une forme hyperbolique de 0,1 mm de diamètre ... 64 Figure 3.5: Intensité relative des ions en fonction leur endroit d’arrivée sur le plan ... 64 Figure 3.6: Effet d’une lentille électrostatique à 800 V sur des ions de charge positive unitaire ayant une énergie initiale de 100 eV ... 66 Figure 3.7: Effet d’une lentille de 20 mm de diamètre à une tension de 800 V à la pression atmosphérique sur des ions de charge positive unitaire ayant une énergie initiale de 100 eV. ... 66 Figure 3.8: Pointe utilisée dans nos simulations. ... 68 Figure 3.9: Faisceau ionique dans une configuration pointe-plan pour diverses pressions. ... 69 Figure 3.10: a) Anneaux de cuivre utilisé pour nos lentilles de type annulaires. b) Lentilles électrostatiques faites par Kimball Physics. ... 72 Figure 3.11: Densité normalisée pour des situations où une lentille électrostatique se situe à 5 mm du plan. ... 73 Figure 3.12: Densité normalisée mesurée au plan pour une lentille de 16,2 mm de diamètre placée à différents endroits dans l’espace inter-électrodes. ... 75 Figure 3.13: Lentille de 16,2 mm de diamètre à divers endroits par rapport au plan... 76 Figure 3.14: Densité normalisée au plan pour des situations où il y a des lentilles électrostatiques multiples dans l’espace inter-électrodes. ... 78 Figure 3.15: Différentes configurations avec des lentilles de 8,1 mm de rayon : (1) lentille à 10 mm du plan, (2) lentille à 7,5 mm du plan et (3) lentilles à 7,5 mm, 5 mm et 2,5 mm. . 79 Figure 4.1: Différents éléments du montage fixés à une table optique. ... 84 Figure 4.2: a) Anneau de cuivre utilisé pour nos lentilles de type annulaires. b) Lentilles électrostatiques faites par Kimball Physics. ... 85 Figure 4.3: Les détecteurs de courants utilisés pour le montage expérimental. ... 86 Figure 4.4: Schéma global du montage ... 87 Figure 4.5: Courbe de la puissance lumineuse normalisée en fonction de la tension appliquée aux bornes de la DEL. ... 89
xi
Figure 4.6: Relation entre la tension prise aux bornes d’une résistance en série avec le phototransistor (module récepteur) et la tension qui alimente la diode émettrice (module émetteur). ... 91
Figure 4.7: Circuit électronique du module émetteur ... 93
Figure 4.8: Connecteur pour joindre les DEL et les phototransistors à la fibre optique. ... 95
Figure 4.9: Circuit électronique du module récepteur ... 96
Figure 4.10: 1ère étape de l’ampèremètre ... 97
Figure 5.1: Relation entre la tension prise aux bornes d’une résistance en série avec le phototransistor (module récepteur) et la tension qui alimente la diode émettrice (module émetteur). ... 102
Figure 5.2: Courant produit par décharge couronne mesuré avec l’ampèremètre et les deux détecteurs. ... 104
Figure 5.3: Pointe utilisée dans le montage : vue de face et vue de côté. ... 106
Figure 5.4: Rapport courant-tension à la pointe en fonction de la tension. ... 107
Figure 5.5: Courants mesurés sur les détecteurs 1 et 2 en fonction de la tension appliquée sur une lentille annulaire de 36,8 mm de diamètre, pour une tension constante à la pointe. ... 109
Figure 5.6: Rapport entre le courant mesuré sur le détecteur 1 et la somme des courants mesurés en fonction de la tension de la lentille, pour une tension constante à la pointe. ... 110
Figure 5.7: Courants mesurés sur les détecteurs 1 et 2 en fonction de la tension appliquée sur la lentille, pour un courant constant à la pointe. ... 111
Figure 5.8: Rapport du courant mesuré au détecteur 1 sur la somme des courants en fonction de la tension de la lentille, pour un courant constant à la pointe. ... 112
Figure 5.9: Lentille de 19,1 mm de diamètre à 10 mm du plan. Courant mesuré sur les deux détecteurs en fonction de la tension appliquée sur la lentille. ... 116
Figure 5.10: Courants mesurés sur le détecteur en fonction de la tension appliquée sur la lentille de 19,1 mm de diamètre pour différents emplacements entre la pointe et le plan. ... 117
Figure 5.11: Courant mesuré sur le détecteur en fonction de la tension appliquée sur la lentille 9,6 mm pour différents emplacements entre la pointe et le plan. ... 118 Figure 5.12: Comparaison des courants mesurés pour la lentille de 9,6 mm de diamètre lorsqu’elle est située à 2,5 mm du plan a), et à 5 mm du plan b) ... 119 Figure 5.13: Lentilles de 15,9 mm, 12,7 mm et 9,6 mm de diamètre à 2,5 mm et 5 mm du plan lorsqu’elles sont à une tension de 2800 V et que la pointe est à une tension de 7000 V. Distribution linéaire de 29 ions espacés de 0,5 mm à 9,96 mm du plan. ... 121 Figure 5.14: Les figures a) et b) sont des agrandissements de l’extrémité gauche de la lentille de 9,6 mm de diamètre telle que présenté à la figure 5.13 d). a) Situation où la pointe est à une tension de 0 V et la lentille de 9,6 mm est à 4400 V. b) Situation où la pointe est à une tension de 7000 V et la lentille de 9,6 mm à 3200 V. ... 123 Figure 5.15: Courants mesurés sur les détecteurs provenant de la lentille de 9,6 mm de diamètre lorsqu’elle se situe à 5 mm du plan. ... 124 Figure 5.16: Lentille cylindrique de 22,9 mm positionnée entre la pointe et le plan dans le montage expérimental. ... 127 Figure 5.17: Courant mesuré sur la cible lorsque les lentilles se situent à 10 mm du plan. ... 128 Figure 5.18: Courant mesuré sur la cible lorsque les lentilles se situent à 7,5 mm du plan. ... 128 Figure 5.19: Courant mesuré sur la cible lorsque les lentilles se situent à 5 mm du plan. 129 Figure 5.20: Courant mesuré sur la cible lorsque les lentilles se situent à 2,5 mm du plan. ... 129 Figure 5.21: Sommaire présentant le courant mesuré sur la cible lorsque toutes les lentilles ont une tension de 2800 V en fonction du diamètre de celle-ci et de l’endroit où elles se situent. ... 131 Figure 5.22: Lentilles 19,1 mm, 12,7 mm et 4,7 mm à 2700 V chacune, espacées de 4,6 mm et à 2,5 mm du plan. La pointe est à 7000 V ... 135
xiii
Liste des tableaux
Table 3.1: Différences dans la densité mesurée sur le plan par rapport à la densité initiale pour différentes pressions. ... 70 Table 5.1 : Caractéristiques des lentilles utilisées ……… 114
xv
Liste des symboles mathématiques
A Fragment moléculaire d’une molécule du gaz échantillon (M)
B Fragment moléculaire d’une molécule du gaz échantillon (M)
d Espacement entre la pointe et le plan
D Coefficient de diffusion
e Charge électrique
e- Électron
eV Énergie d’un électron accéléré par une différence de potentiel de 1 V
E Champ électrique
Ei Énergie d’ionisation
h Longueur du pas entre chaque noeud
I Courant
IC Courant dans la borne collectrice d’un transistor
IE Courant dans la borne émettrice d’un transistor
j Densité de courant
j0 Densité de courant maximale
J Flux d’ions
kB Constante de Boltzmann
K Coefficient de mobilité ionique
L Longueur du parcours d’un ion
masseion Masse de l’ion
massegaz Masse du gaz neutre
M Molécule du gaz échantillon
M+ Molécule ionisée positivement
M- Molécule ionisée négativement
n Densité de particules
ni Nombre d’ions
nstats Nombre de collisions statistiques compilées
n0 Nombre d’atomes neutres
p Pression
rion Déplacement de l’ion
rstats Saut de longueur aléatoire
R Résistance
RL Résistance de charge
RV Résistance variable
t Intervalle de temps
T Température
vd Vitesse moyenne de dérive
vt Vitesse thermique du gaz
vion Vitesse moyenne des ions
V Tension
VC Tension dans la borne collectrice d’un transistor
VE Tension dans la borne émettrice d’un transistor
Vseuil Tension seuil de la diode
Vsp Tension de claquage de la décharge couronne
V0 Tension d’allumage de la décharge couronne
X Molécule d’un gaz réactif
W Fonction de travail
E/n Champ électrique réduit
E/p Rapport du champ électrique sur la pression
α Premier coefficient de Townsend
ε0 Permittivité du vide
θ Angle radian
λ Libre parcours moyen
xvii ρ Distribution de charge μ Masse réduite Ω Section efficace n ou dn/dx Gradient de concentration
xix
Remerciements
La présentation de ce mémoire est l’accomplissement d’un travail que je n’aurais pu réussir sans l’aide de plusieurs personnes. Dans un premier temps, je remercie mon co-directeur de maîtrise, le professeur Réal Paquin qui fût pour moi un guide et un mentor. Par son enthousiasme et son grand courage malgré des moments plus difficiles, il m’a permis de compléter cette importante étape. Sa patience et ses connaissances sans limites m’ont aidé dans le cheminement ardu que peut être un tel projet. Par son entremise j’ai eu la chance de collaborer avec une équipe géniale, nommé, Phytronix Technologies. Grace à leur soutien financier, technique et moral, j’ai pu achever ma maitrise. C’est avec joie que j’ai donc décidé de poursuivre mon parcours académique vers un projet de doctorat.
Je veux également remercier mon autre co-directeur, le professeur Simon Rainville pour avoir accepté de co-dirigé ma maîtrise. Ses précieux conseils dans les moments où j’en avais besoin m’ont permis d’accomplir ce projet.
La réalisation d’un aussi long projet nécessite un équilibre entre la vie académique et la ‘’vraie’’ vie. À ce titre, Pascal, Kevin, Tyler et plusieurs autres amis se sont chargés de me changer les idées plus souvent que j’aurais pu le souhaiter en venant passer quelques jours à Québec. Pour me ressourcer, je pouvais également compter sur ma famille qui m’offrait toujours un oasis de paix à Gatineau lorsqu’il a pu faire tempête. Enfin, je tiens à remercier ma copine Myriam pour sa patience, son encouragement ainsi que son support essentiel pendant la rédaction de ce document.
Un gros merci à Louis Harbour et Daniel Landry avec lesquels je pouvais toujours avoir des discussions intéressantes et constructives sur nos projets et sur la physique en général. Plus d’une fois, ils m’ont dépanné et aidé avec des problèmes mathématiques.
Finalement, les nombreux étudiants d’été québécois ou français ont amené beaucoup de vie pendant ces quelques années passées au LPAM.
Merci à tous ceux que je puis côtoyer pendant ces dernières années pour le support que vous avez pu m’apporter.
1
Chapitre 1 : Introduction
1.1. Historique de la spectrométrie de masse et but du projet
Le spectromètre de masse est aujourd’hui l’instrument analytique le plus répandu et utilisé dans le monde pour l’identification des produits chimiques 1. Le premier à avoir conçu un appareil digne de ce nom est l’Anglais Joseph John Thomson du Laboratoire Cavendish de l’Université de Cambridge au début des années 1900 2. Son étudiant F. W. Aston perfectionna le concept de façon à créer un appareil beaucoup plus performant. Presque simultanément, en Amérique, A. J. Dempster de l’Université de Chicago créa aussi un spectromètre de masse ayant une géométrie différente de ceux créés au Laboratoire Cavendish. À partir de ce moment, la spectrométrie de masse était lancée.
On peut affirmer que deux concepts ont mené les scientifiques vers la conception du spectromètre de masse (MS). Dans un premier temps, Sir Williams Crookes exprima l’idée qu’il pourrait exister des atomes de la même espèce ayant un poids atomique différent : ce que nous appelons maintenant les isotopes. La découverte d’éléments radioactifs prouva la validité de cette hypothèse. Ailleurs, deux Allemands firent l’observation de rayons anodiques. Eugene Goldstein observa un rayonnement lumineux à partir de la cathode lors de décharges gazeuses à basse pression. Puis, Wilhelm Wien démontra que ces rayons pouvaient être déviés par des champs magnétiques forts; ce qui confirma que les rayons anodiques étaient des faisceaux de particules chargées positivement.
J. J. Thompson est parti du concept des rayons anodiques pour créer un appareil dans lequel les champs magnétiques et électriques intenses étaient superposés permettant aux ions de parcourir des trajectoires paraboliques spécifiques selon leur rapport m/z (m étant
la masse de l’ion et z sa charge). Ainsi, il était capable de séparer la masse de plusieurs gaz avec une résolution de moins de 10%. Pour Thomson, cette nouvelle technique d’analyse avait l’avantage, non seulement de nécessiter une petite quantité de matériel, mais également d’ouvrir les portes à résoudre des problèmes d’identification chimique avec une plus grande facilité que tout ce qui existait à ce moment-là: «I feel sure that there are many problems in chemistry which could be solved with far greater ease by this than by any other method» 1. Sans le savoir, il fût le premier à observer des isotopes (de néon) au moyen d’un spectromètre de masse. L’importance de l’avènement du spectromètre de masse est primordiale dans ce domaine, car les isotopes sont très difficilement dissociables chimiquement et la différence de masse, somme toute très petite, demeure le meilleur moyen de les mesurer.
Les grandes perspectives qu’avançait Thomson ont amené plusieurs personnes à travailler sur le concept. Un des personnages majeurs fût Francis William Aston, assistant de recherche de J. J. Thomson au Laboratoire Cavendish. Dans le but d’améliorer la résolution de l’instrument sans altérer l’intensité au détecteur 2, il choisit de séparer la section en deux parties distinctes où les ions sont soumis aux champs électriques et magnétiques. Les ions, désormais focalisés par un système de lentilles électrostatiques, passent entre deux électrodes planes qui produisent un champ électrique. Dans cette première section, les ions sont séparés selon leur énergie cinétique, ce qui n’était pas possible avec l’instrument de Thomson. Dans la deuxième section les ions trouvent un champ magnétique qui fait converger les ions de même masse sur une plaque photographique placée au plan focal. L’apport majeur de cette géométrie est que les ions étaient maintenant focalisés en un seul endroit pour un petit spectre d’énergie. Avec son instrument, il put répertorier un nombre important d’éléments ayant des isotopes.
Dans les mêmes années, à l’Université de Chicago, Arthur Jeffrey Dempster inventa un spectromètre de masse à secteur magnétique de 180 qui faisait converger les ions, de même masse, vers un point focal. Son concept était différent d’Aston par le fait qu’il
3 n’avait pas de filtre d’énergie; il accélérait plutôt les ions pour rendre le faisceau mono-énergétique. Ensuite, les ions étaient introduits par une fente dans un champ magnétique qui faisait 180. La divergence angulaire du faisceau ionique limitée par la fente d’entrée était approximativement compensée au second degré. Son instrument lui permit de découvrir de nouveaux isotopes et inspira d’autres chercheurs dans la conception d’appareils plus performants.
Les spectromètres de masses étaient surtout utilisés en milieu académique et ils étaient de conception artisanale jusqu’au début des années 1940. À ce moment, l’industrie pétrolière commença à utiliser l’instrument à des fins de prospection et d’analyse de produits raffinés 3. La compagnie Consolidated Engineering Corporation fût la première à commercialiser un spectromètre de masse en 1943. La Deuxième Guerre mondiale donna un essor formidable au domaine, car, pour les besoins du projet Manhattan, les scientifiques utilisèrent ce moyen pour mesurer précisément le ratio d’uranium U235/U238 et pour collecter les ions fissibles. C’est d’ailleurs en raison de cette utilisation qu’on utilise le terme collecteur lorsqu’on parle de détecteur.
On peut diviser le spectromètre de masse en trois parties distinctes : la source d’ionisation, l’analyseur et le détecteur. Depuis le début de cette science, les chercheurs mirent beaucoup d’efforts dans le but d’améliorer la partie analyseur. Une première gamme d’appareils de type statique fut construite. Dans ce type d’instrument on retrouve les systèmes ayant plusieurs secteurs magnétiques et qui peuvent aussi avoir des lentilles électrostatiques. Puis une deuxième gamme de spectromètres de type dynamique vit le jour. Ces appareils étaient dynamiques par le fait que la détermination de la masse des ions était fonction du temps. Les spectromètres de masse à temps de vol (TOF) ainsi que ceux fonctionnant en résonance cyclique sont de ce type. Aujourd’hui, on retrouve presque qu’exclusivement des spectromètres de masse dynamique sur le marché. De nouvelles applications se développèrent par la suite en même temps que de nouvelles géométries étaient inventées et testées par les chercheurs.
Les autres parties du spectromètre de masse connurent aussi une évolution. Au niveau du détecteur, on utilisait, dans les débuts, une plaque fluorescente qui émettait des photons lorsqu’un ion la percutait, mais qui ne gardait aucune trace après l’impact. Ensuite, on utilisa la plaque photographique que le faisceau d’ions marquait lorsqu’il la heurtait. Puis, Dempster fut le premier à utiliser un détecteur électronique avec lequel on mesurait le courant ionique reçu. Ce type de détecteur, connu aussi sous le nom de cylindre de Faraday, est devenu la norme par la suite. Comme le raffinement de la technique pour permettre une meilleure résolution au niveau de la masse diminuait l’intensité des ions, des moyens pour amplifier le signal mesuré ont dû être développés. On a d’abord amplifié le courant directement, puis les multiplicateurs d’électrons apparurent pour permettre des progrès majeurs de détection lorsque le courant ionique est très faible.
La source d’ionisation a aussi subi des développements avec le temps. Selon Roboz: «the functions of an ion source are to produce as many ions as possible from the neutral particles present and to form, shape, and eject an ion beam that is suitable to entrance into the analyser» 2. Cette section du spectromètre de masse a reçu des améliorations constantes depuis les débuts à l’instar des deux autres sections. Encore aujourd’hui, une grande partie des efforts mis dans l’amélioration de la technique se fait au niveau de la source. C’est cette partie qui nous intéressera tout particulièrement dans ce mémoire.
Les méthodes d’ionisation divergent grandement et ont toutes leurs avantages et leurs désavantages. Dans les appareils de Thomson et Aston, l’ionisation était produite par des décharges électriques dans un tube maintenu à basse pression. Cette source n’était pas optimale, car même si un grand courant ionique était produit, les ions entraient dans l’analyseur avec un large spectre d’énergie 2-4. En utilisant toujours le concept de décharge gazeuse à basse pression, certains firent en sorte que les ions extraits étaient mono-énergétiques, mais, en somme, le courant restait très instable. L’étalement en
5 énergie (eV) des ions, lorsqu’ils sont produits, est une des caractéristiques les plus importantes dont on doit tenir compte 2. Plus l’étalement est réduit, plus il est possible de faire converger facilement les ions vers un point précis. C’est ainsi que les chercheurs se tournèrent vers d’autres façons d’ioniser la matière. On vit l’apparition de sources utilisant l’ionisation par plasma, par bombardement d’électrons, par décharge RF, par effets de champ, etc. Toutes ces techniques requièrent une géométrie et une connaissance différente l’une de l’autre, ce qui rend les améliorations difficiles. De plus, aucune de ces techniques n’a permis de produire une source universelle permettant d’ioniser tous les composés avec autant d’efficacité. Il est très important dans le domaine de la spectrométrie de masse de savoir ce qu’on veut analyser pour pouvoir choisir la source, l’analyseur et le détecteur qui nous sied le mieux. Comme l’analyseur doit être maintenu sous vide pour pouvoir différencier les masses, les sources ont été conceptuellement pensées pour fonctionner elles aussi sous vide (10-2 – 10-8 Torr). Cependant, l’insertion des échantillons à analyser était particulièrement ardue et lente, pour ne pas altérer le vide du spectromètre de masse.
Les chimistes développèrent une puissante technique leur permettant de séparer les composés chimiques en phase gazeuse en les faisant passer dans une colonne ayant une phase liquide stationnaire. On appela cette technique la chromatographie en phase gazeuse (GC). La technique ne peut être utilisée qu’avec des composés volatils. À la sortie de la colonne les molécules sont ionisées par différentes techniques, puis le courant ionique produit est détecté. Pour raffiner l’analyse des composés chimiques, on a voulu coupler le chromatographe en phase gazeuse avec un spectromètre de masse (GC-MS), pour former un super-détecteur. Le même cheminement se fit avec la technique de la chromatographie en phase liquide (LC) aussi appelé HPLC (High Pressure Liquid Chromatography), car elle nécessite une grande pression à l’injection du liquide dans la phase solide. Elle fût aussi couplée à un spectromètre de masse (LC-MS). Le couplage, autant pour les phases gazeuse et liquide, obligèrent les chercheurs à développer de nouvelles interfaces d’entrée pour les spectromètres de masse ainsi que de nouvelles
sources d’ionisation, car la sortie du chromatographe se faisait à la pression atmosphérique. La chromatographie en phase gazeuse ou liquide couplée avec un spectromètre de masse (GC-MS, LC-MS) est une technique puissante qui est encore largement utilisée dans le domaine.
L’avènement des chromatographes en phase liquide couplés à un spectromètre de masse amena les scientifiques à créer des sources opérant à la pression atmosphérique. Cependant, à cette pression, l’ionisation est beaucoup plus difficile et les courants produits sont très inférieurs comparativement à ceux produits sous vide. Parmi les différentes techniques employées, se retrouvent l’ionisation par désorption laser, l’ionisation chimique, l’électro-nébulisation, la nébulisation thermique et l’ionisation par décharge couronne. Un des avantages à produire des ions à la pression atmosphérique est que par le biais de nombreuses collisions occasionnées par une plus grande densité du gaz, les analytes ionisés n’acquièrent pas l’énergie nécessaire à leur fragmentation. On a donc pu mesurer des molécules de plus en plus massives. Maintenant, les sources à la pression atmosphérique sont largement utilisées pour travailler avec des protéomes, de l’ADN, des lipides et tout autre composé d’une très grande masse et nécessitant d’être ionisé doucement. Ainsi, dans les vingt dernières années, des sources se nommant MALDI (Matrix-Assited Laser Desorption/Ionization), DESI (Desorption Electrospray Ionization), LDTD (Laser Diode Thermal Desorption) et bien d’autres ont vu le jour. Ce qui était au début un appareil servant à répertorier les différents éléments du tableau périodique et leurs isotopes est donc devenu un instrument d’analyse universel avec lequel on peut analyser des composés chimiques ayant des poids variant de quelques masses atomiques à des dizaines de milliers d’unité de masses atomiques. Comme toute amélioration à l’une des trois parties d’un spectromètre de masse est non négligeable, nous avons décidé d’étudier le phénomène d’ionisation et le transfert d’ions de la source vers le spectromètre de masse, plus particulièrement dans le cas de la décharge couronne utilisée dans la source LDTD fabriquée par Phytronix Technologies.
7 La source LDTD désorbe les analytes thermiquement par un faisceau en proche infrarouge produit par une diode laser, puis ceux-ci sont amenés par un gaz vecteur vers une pointe maintenue à un très haut potentiel (~5000V) qui les ionise par décharge couronne 10. Par la suite, les ions créés dérivent, dans le champ électrique, vers l’entrée d’un spectromètre de masse. La décharge couronne produite dans une configuration pointe-plan émet un faisceau ionique qui diverge grandement selon l’importance de la distance entre la pointe et le plan. C’est ainsi qu’on a émis l’hypothèse qu’en modifiant le champ électrique ( E) dans la région séparant la pointe du plan nous pourrions produire un effet de convergence sur les ions. Le but du projet est donc d’obtenir la focalisation d’un faisceau ionique dans une configuration pointe-plan par l’utilisation de lentilles électrostatiques.
1.2. Objectifs du projet et méthodologie
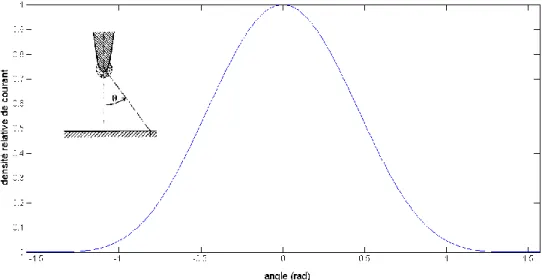
Le but du projet est de réussir à focaliser un faisceau ionique à la pression atmosphérique. On peut produire des ions dans les deux polarités avec la décharge couronne 9. Pour simplifier le montage et le nombre d’expériences à réaliser, nous nous sommes limités à travailler avec des ions de polarité positive. On sait que la décharge couronne produit un courant relativement stable de quelques microampères lorsqu’on y applique un potentiel de quelques kV sur une distance d’environ 1 cm. Le fonctionnement et les résultats obtenus avec la source LDTD prouvent que ce phénomène ionise assez d’analytes pour réaliser l’analyse par spectrométrie de masse 5. Comme il a été mentionné précédemment, la configuration pointe-plan favorise une grande dispersion des ions. La densité de courant ionique
J
mesuré sur le plan se comporte selon la loi empirique
de Warburg (1.1) 6, 7, 8 trouvée selon des expériences faites il y a plus de 110 ans (figure1.1) et qui a comme forme :
5 0
( ) cos
j j (0.1)
où θ est l’angle par rapport à la pointe et j0 représente le courant ionique à son maximum
(au centre).
Figure 1.1: Densité relative du courant ionique en fonction de l'angle (rad)
On constate que la densité du courant devient très faible lorsque l’angle atteint 1,5 radian. On peut aussi observer qu’une grande densité de courant est dispersée autour de θ = 0. Pour un espacement pointe-plan de 10 mm, on peut mesurer du courant jusqu’à 17,3 mm du centre. Comme l’entrée d’un spectromètre de masse a un rayon de l’ordre du millimètre, on réalise qu’une grande partie du courant produit est perdue pendant le transfert vers le spectromètre de masse. Notre hypothèse est qu’en modifiant la géométrie du champ électrique dans l’espace séparant la pointe de l’entrée du spectromètre de masse, on pourrait augmenter le courant ionique y entrant. Pour y parvenir, on a choisi d’utiliser des lentilles électrostatiques à symétrie cylindrique par souci de simplicité. Le projet est exploratoire, car il n’y a pas réellement de résultats avancés dans la littérature faisant référence à des électrodes placées dans l’espace pointe-plan. En allant perturber le champ à la pointe, nous risquons de le modifier et d’altérer le phénomène d’ionisation. Toutefois, le transport d’ions à la pression atmosphérique est
9 très largement dominé par le champ électrique. La question est de savoir si on peut modifier avantageusement le champ électrique de façon à focaliser le faisceau ionique dans une configuration qui tend naturellement à le faire diverger.
Pour répondre à cette question, nous avons cru bon d’étudier théoriquement le phénomène d’ionisation par décharge couronne, la mobilité ionique ainsi que le transport d’ions. Cette étude a pour but de caractériser le phénomène et ses limites afin de pouvoir exploiter le courant produit à son maximum. Comme le champ électrique a un effet prédominant, nous voulons utiliser un logiciel permettant de visualiser celui-ci. Dans le but d’observer le phénomène expérimentalement, un montage est construit pour mesurer le courant ionique.
Le mémoire débute par un chapitre présentant une revue théorique des différentes façons d’ioniser, dans le vide ainsi qu’à la pression atmosphérique, en approfondissant particulièrement la décharge couronne. La mobilité ionique et le transport ionique sont aussi abordés, ainsi qu’un aperçu sur les différentes expériences réalisées avec des lentilles électrostatiques. Le chapitre suivant est consacré aux simulations numériques. On y explique les techniques utilisées pour résoudre l’équation de Laplace, puis les logiciels de simulation utilisés sont présentés. On expose ensuite nos résultats et analyses sur les simulations réalisées. Le quatrième chapitre décrit le montage expérimental en le divisant en trois parties distinctes : mécanique, électronique et numérique. L’étalonnage ainsi que les limites de notre système y sont exprimés. Les résultats et analyses obtenus avec notre montage sont posés dans le cinquième chapitre. Le dernier chapitre conclut en faisant un survol des résultats obtenus dans les différentes parties du mémoire.
11
Chapitre 2 : Revue des technologies et concepts
2.1 Introduction
Le but du projet est de réaliser un effet focalisant sur un faisceau ionique. Nous nous sommes intéressés à cette problématique car ce type de faisceau est utilisé dans certaines sources ioniques à la pression atmosphérique. Il est important de produire un faisceau ionique qui est optimal pour entrer dans un spectromètre de masse. Pour ce chapitre et les subséquents, on appellera luminosité de la source, la partie du faisceau ionique entrant dans le spectromètre de masse. Dans le cas de la décharge couronne utilisée pour faire de l’ionisation chimique à la pression atmosphérique (APCI), technique utilisée dans la source LDTD entre autres, la configuration pointe-plan produit un faisceau ionique grandement divergent. La luminosité d’une telle source est donc faible. La focalisation du faisceau permettrait d’augmenter la luminosité de la source et pourrait permettre ultimement d’augmenter le rapport signal sur bruit dans le spectromètre de masse. De plus, en chimie analytique, on travaille souvent à l’état de traces et une amélioration quantitative du signal ne peut être que bénéfique.
L’ionisation à la pression atmosphérique et la recherche d’efficacité du transfert d’ions de la source vers le spectromètre de masse a débuté avec l’avènement de la chromatographie en phase gazeuse et liquide. La découverte de ces techniques permit aux scientifiques de séparer les divers éléments d’un composé chimique pour en faire une étude qualitative. Les spectromètres de masse permettent difficilement de travailler avec des mélanges complexes, car les spectres produits deviennent rapidement indéchiffrables. Cependant, la spectrométrie de masse permet une étude quantitative avantageuse. Les deux techniques ont donc été couplées pour former ce qu’on peut appeler un super
détecteur. Le couplage d’un chromatographe en phase gazeuse ou liquide avec un spectromètre de masse a amené la modification de l’interface d’entrée de celui-ci.
Le problème majeur vient du fait que le flux sortant d’une colonne de chromatographe est à une pression plus élevée que la pression de fonctionnement d’un spectromètre (10-6 – 10-7 Torr). Le flux a donc tendance à avoir une expansion qui disperse les composés que l’on veut analyser. Pour permettre de coupler efficacement un chromatographe et un spectromètre de masse, des interfaces d’entrée ont été conçues. Parmi celles-ci ont retrouve l’interface par membrane dite sélective, car elle laisse passer seulement les composés désirés vers le spectromètre de masse. L’introduction directe est une autre méthode qui consiste à tout simplement introduire la colonne du chromatographe dans l’entrée du spectromètre de masse 12. Le jet moléculaire supersonique est une interface utilisée en chromatographie en phases gazeuse et liquide, ainsi que pour les sources à la pression atmosphérique. Ce jet est produit à l’aide d’une interface située entre deux régions de pression différente qui a un orifice sonique 15. Cet orifice permet à un gaz provenant d’une région à plus haute pression de subir une expansion adiabatique de l’autre côté. Cette expansion garde l’énergie des analytes constantes, mais la température du gaz diminue, ce qui entraine une vitesse plus élevée pour les particules. L’expansion entraine les particules plus légères (souvent les solvants par rapport aux analytes) à se disperser sur les côtés et les particules plus lourdes à rester au centre. Ceci a pour effet de concentrer les particules à analyser au même endroit. L’entrée du spectromètre de masse a donc été placée au centre du jet moléculaire supersonique. D’autres interfaces ont été fabriquées de façon à optimiser l’entrée des analytes dans le spectromètre de masse.
Pour le chromatographe en phase liquide, une autre variable entre en jeu, soit la nature liquide et non-volatile de l’échantillon, ce qui rend encore plus difficile son couplage. Les interfaces ont donc été construites dans le but de faire évaporer les solvants utilisés en chromatographie en phase liquide et les techniques développées pour le GC-MS ont été appliquées ici. Le flux était, par la suite, dirigé vers une source utilisant l’impact
13 électronique ou l’ionisation chimique qui opèrent sous vide (10-3 – 10-5 Torr). Tous ces développements guidèrent les chercheurs vers des solutions applicables à la pression atmosphérique. Les sources d’ionisation à la pression atmosphérique furent inventées pour permettre le couplage des chromatographes avec le spectromètre de masse, mais leur utilisation a été élargie à beaucoup d’autres applications.
Maintenant que nous connaissons l’origine de l’ionisation à la pression atmosphérique, nous nous intéresserons à l’ionisation proprement dite. En premier lieu, un bref survol sera fait des méthodes d’ionisation utilisées sous vide, car elles sont, de façon détournée, utilisées pour l’ionisation à la pression atmosphérique. Puis, on étudiera les différentes sources d’ionisation à la pression atmosphérique, en particulier la source LDTD. Par la suite, la théorie de la décharge couronne sera approfondie. Un sommaire sera fait des études ayant pour sujet les lentilles électrostatiques à la pression atmosphérique. En dernier lieu, la mobilité ionique sera abordée pour mieux comprendre la contrainte que la pression atmosphérique nous impose.
2.2 Les méthodes d’ionisation utilisées dans le vide
Il est important d’examiner les différentes méthodes d’ionisation à basse pression à l’origine des premières sources ioniques, car elles ont été intégrées, de manière diverses et améliorées, dans la fabrication de sources à la pression atmosphérique. On résumera ici cinq méthodes employées dans le vide qu’on retrouve aussi à la pression atmosphérique : l’ionisation par bombardement électronique, effet de champ, par création d’un plasma ainsi que l’ionisation chimique et l’ionisation de surface (thermique). Ces types d’ionisation furent largement utilisés dans les débuts de la spectrométrie de masse. La grande luminosité des sources sous vide (10-1 – 10-10 A) demeure le grand avantage par rapport à l’ionisation à la pression atmosphérique. Cette grande luminosité était cependant indispensable, car les détecteurs et analyseurs du moment n’étaient pas aussi
performants qu’aujourd’hui. La qualité du vide varie selon la méthode employée (1 – 10-8 Torr) 4.
2.2.1 L’ionisation par bombardement électronique
L’ionisation par bombardement électronique, aussi appelé impact électronique, ionise un gaz électroniquement neutre par collisions d’électrons avec celui-ci. Pour obtenir l’ionisation d’un gaz (M), un faisceau d’électrons doit être accéléré de façon à avoir une énergie cinétique plus élevée que l’énergie d’ionisation du gaz. L’ionisation des particules neutres (équation 2.1) n’est qu’un des phénomènes parmi plusieurs qui peuvent se produire lorsqu’on bombarde un gaz avec des électrons. Le faisceau d’électrons peut provoquer la fragmentation (A, B) des molécules (équation 2.2), l’excitation des atomes/molécules ou simplement subir des collisions élastiques. Il peut se former des ions négatifs (équation 2.3), mais ceux-ci ne sont considérés que pour quelques substances.
2 M e M e (2.1) 2 M e AB e (2.2) M e M (2.3)
Ces phénomènes arrivent tous dans des proportions différentes selon l’énergie des électrons incidents. Lorsque les électrons sont peu énergétiques (moins que l’énergie d’ionisation), il est plus probable que les atomes soient excités ou que l’électron diffuse carrément. Lorsque l’énergie est assez grande, un électron peut produire plusieurs ions, chargés simplement ou de façon multiple, tout au long de son parcours. Des relations décrivant précisément le courant ionique produit en fonction du nombre d’électrons incidents sont connues et se retrouvent dans la littérature 2,4.
15 L’ionisation étant plus efficace aux environs de 70 à 150 eV (figure 2.1) pour l’ensemble des gaz mesurés, les sources utilisant cette technique accélèrent le faisceau d’électrons au niveau de ces énergies. Ce type de source fonctionne bien avec entre autres, des gaz, des vapeurs de composés organiques et des vapeurs de solides. Comme il a été mentionné précédemment, les électrons peuvent fragmenter les molécules, ce qui peut être un avantage lorsqu’on veut étudier la composition interne des molécules.
Figure 2.1: Efficacité d'ionisation selon l'énergie des électrons 71
Un filament de tungstène dans lequel passe un grand courant est utilisé pour émettre les électrons. Le faisceau d’électrons est dirigé dans une enceinte close où il interagit avec le gaz ambiant. Les ions formés sont, quant à eux, extraits vers le spectromètre de masse perpendiculairement au faisceau d’électrons avec des électrodes focalisantes. Pour améliorer l’efficacité d’ionisation par électrons, certaines sources allongent le parcours
des électrons avec un champ magnétique leur faisant poursuivre leurs trajectoires en forme de spirale ou en faisant tout simplement osciller les électrons entre deux électrodes négatives. La source ionique par bombardement d’électrons opère sous un vide de ~10-4 Torr et fournit un courant ayant un étalement en énergie restreint et stable (~ 10 eV) 4.
D’autres techniques ont vu le jour en utilisant des atomes ou des ions à la place d’électrons pour ioniser. Ceci donna l’ionisation par jet rapide d’atomes (Fast atom bombardment FAB), et l’émission d’ions secondaires (Secondary ion mass spectrometry SIMS).
2.2.2 L’ionisation chimique
L’ionisation chimique ressemble beaucoup à l’ionisation par bombardements d’électrons, mais s’avère plus douce pour ioniser. De plus, on opère une source utilisant ce type d’ionisation à une pression supérieure à l’ionisation par impact électronique (10-2 Torr). Un faisceau d’électrons est utilisé pour ioniser un gaz dit réactif (CH4, H2, NH3) comme cela a été présenté à la section précédente (2.2.1). L’échantillon à analyser est mélangé en très faible quantité dans le gaz réactif (X). La pression étant plus élevée que pour l’impact électronique, les ions collisionnent avec les molécules neutres du gaz réactif et, dans une plus faible proportion, avec les molécules neutres de l’échantillon (M). Ces nombreuses collisions subies par les ions peuvent modifier leur composition. L’ionisation positive des molécules neutres se fait majoritairement par deux mécanismes soit l’échange de charge ou le transfert d’un proton 13. L’échange de charge (équation 2.4) se produit lorsqu’un ion entre en collision avec une molécule du gaz ambiant (M ou X). Si l’ion a une assez grande énergie (> que l’énergie d’ionisation Ei de M ou X), il peut arracher un électron à une molécule stable et former un ion. Le transfert de proton (équation 2.5), est le mécanisme dominant pour l’ionisation chimique 11. Celui-ci consiste en ce que les
17 molécules ionisées du gaz réactif (X) accumulent un proton de plus au courant de leurs collisions avec les molécules neutres réactives et les donnent aux molécules du gaz échantillon (M).
XM X M
(2.4)
XHM X M H (2.5)
Plusieurs gaz réactifs peuvent être utilisés, mais le gaz échantillon doit être une base plus forte pour attirer un proton. Le méthane est une des bases utilisées et l’équation 2.6 nous montre comment les différentes réactions ion-molécule génèrent un ion positif. L’ionisation chimique négative peut aussi se produire lorsque le gaz échantillon perd un proton comme lorsque le gaz réactif favorise la formation de OH- (équation 2.7)
4 4 4 4 5 3 5 4 2 CH e CH e CH CH CH CH CH M CH M H (2.6)
2 OHM M H H O (2.7)L’ionisation chimique est dite plus douce que l’ionisation par impact électronique, car l’énergie du gaz réactif est beaucoup moindre que celle du faisceau d’électrons, ce qui tend à ioniser les molécules neutres sans les fragmenter. Cette technique est prisée lorsqu’on ne veut pas dénaturer le composé étudié.
2.2.3 L’ionisation de surface
L’ionisation de surface est aussi appelée l’ionisation thermique. Le principe est qu’en chauffant une surface où l’échantillon a été absorbé, celui-ci se désorbera sous la forme d’ions ou d’atomes/molécules neutres. On peut déposer préalablement l’échantillon sur la surface froide et, par la suite, la chauffer ou simplement diriger des vapeurs de l’échantillon sur la surface déjà chauffée. Classiquement, une surface conductrice était utilisée et on y faisait passer un grand courant pour la chauffer. Cependant il existe d’autres moyens efficaces de faire monter la température de la surface comme avec un laser. La surface requiert d’avoir une grande fonction de travail (W), ainsi l’iridium, le rhénium, l’osmium, le tantale, le tungstène et le platine sont les éléments le plus souvent utilisés. Les métaux alcalins sont plus susceptibles d’être ionisés positivement, tandis que le groupe des halogènes, qui ont une très grande affinité électronique, créent des ions négatifs. L’ionisation négative est toutefois très difficile. La technique permet d’ioniser des éléments ayant un poids atomique élevé. Le rendement de formation ionique (équation 2.8), soit le rapport d’ions versus les atomes/molécules neutres n ni o, est donné par :
( ) exp( ) i i o n e W E n kT (2.8)
où k est la constante de Boltzmann, e la charge de l’électron, ni le nombre d’ions et n0 le
nombre d’atomes neutres.
Ce rendement de formation ionique dépend beaucoup de la fonction de travail de la surface, mais aussi de la température 2. Plus W est grand par rapport à l’énergie d’ionisation, plus n ni o diminue si la température augmente. Si W est plus petit que Ei, le
rapport n ni o augmente avec la température, mais le nombre total d’ions créés est beaucoup moindre que dans la situation précédente. Il est à noter que les échantillons ont
19 avantage à avoir la plus petite température d’évaporation pour être efficacement ionisés. Cette technique n’a pas tendance à dégrader l’échantillon et permet de prendre des mesures de rapport isotopique très précises. Les sources fonctionnant avec ce type d’ionisation opèrent dans un très grand vide (10-6 Torr) et peuvent produire un courant ionique de 10-2 à 10-5 A qui a un très faible étalement en énergie 4.
2.2.4 L’ionisation par effet de champ
L’ionisation par effet de champ ionise un échantillon lorsqu’on applique un grand potentiel entre deux électrodes. En plaçant une électrode en forme de pointe à un potentiel élevé (5000 V) et une seconde électrode à la masse, on crée les conditions favorables à l’effet tunnel d’électrons de l’échantillon vers la surface conductrice 14. Pour ioniser les analytes, on doit les amener près de la surface. Ainsi, on peut vaporiser l’échantillon, s’il est volatile, ou le déposer préalablement s’il est non volatile. Le champ électrique déforme la barrière de potentiel de l’analyte ce qui permet aux électrons de s’échapper dans la surface conductrice. Les sources à ionisation par effet de champ fonctionnent dans un vide d’environ 10-8 Torr, et produisent des courants faibles (10-10 à 10-8 A) 4. La densité de courant, par contre, est très élevée ce qui est un avantage. L’étalement en énergie des ions créés est beaucoup plus important que pour les techniques présentées précédemment. Enfin, cette technique ionise les analytes doucement sans les fragmenter.
L’ionisation négative peut aussi se produire lorsque le champ électrique produit par une pointe négative permet l’émission d’électrons qui, par la suite, ionisent le gaz ambiant. Cependant, les sources classiques à effet de champ fonctionnaient majoritairement en mode positif.
L’efficacité d’ionisation dépend de la grandeur du champ électrique qui lui-même dépend du potentiel de la pointe et de son rayon de courbure. Plus le rayon de courbure est petit, plus le champ électrique sera intense pour un même potentiel, ce qui aura pour effet d’ioniser un maximum d’analytes. Il est évident que le rayon de courbure est approximatif, i.e. que la surface n’est pas parfaitement lisse, des aspérités peuvent ressortir en certains endroits et produire un champ électrique très élevé localement. Ce phénomène fût à la base du concept de la microscopie par effet de champ où l’on peut observer la surface de l’émetteur de façon très précise.
2.2.5 L’ionisation par plasma
L’ionisation se produit dans un plasma qui s’auto-génère. Le concept est similaire à l’ionisation par bombardements électronique, i.e. que l’échantillon est ionisé par collisions avec des électrons présents dans le plasma. La différence réside dans le fait que les électrons ionisant les analytes ont l’énergie correspondant à la température du plasma et non d’un faisceau d’électrons (electron gun). Le plasma peut être créé dans un volume et un espace moins contraignant que pour bien d’autres techniques. Le plasma doit être initié avant de pouvoir s’auto-générer. Ainsi en bombardant le gaz ambiant, on produit une avalanche d’électrons qui, à leur tour, ionisent et créent d’autres électrons pour enfin atteindre un équilibre. Une électrode de polarité négative est installée de manière à pouvoir extraire les ions.
Le plasma peut être généré de différentes façons et selon diverses configurations d’électrodes. La décharge rf produit un plasma en faisant osciller les électrons à une fréquence de 10 à 30 MHz dans un vide de 10-4 à 10-2 Torr 4. Le momentum acquis par collision, ajouté au changement de direction du champ électrique, donne assez d’énergie aux électrons pour ioniser les analytes tel que décrit par l’équation 2.1. Le fait de faire osciller un électron allonge son parcours ce qui augmente, du même coup, la probabilité
21 qu’il ionise une particule. On peut aussi appliquer un champ magnétique pour faire tourner les électrons dans la cavité. La décharge rf crée des ions ayant une grande divergence en énergie.
L’utilisation d’un plasma pour ioniser se retrouve aussi dans les sources fonctionnant avec des cathodes chaudes ou froides, ainsi que pour les sources plasmatron et duoplasmatron. Toutes ces sources ont une géométrie originale pour permettre une ionisation et une extraction pouvant convenir à différents types d’utilisation.
La décharge luminescente (glow discharge) crée un plasma entre deux électrodes planes dans une enceinte maintenue aux alentours de 1 Torr. Les deux électrodes créent un fort champ électrique qui permet aux électrons et aux ions de dériver dans des directions opposées. Les électrons créent les ions par collision avec le gaz neutre. Cette technique est intéressante, car elle est à la base du concept de la décharge couronne à la pression atmosphérique.
En somme, l’ionisation par plasma propose une méthode d’ionisation versatile pouvant être utilisée dans une configuration qui convient au montage employé.
2.2.6. Résumé
Différentes méthodes d’ionisation dans le vide ont été présentées. On a pu constater que la qualité du vide ainsi que les caractéristiques du faisceau ionique diffèrent grandement d’une technique à l’autre. Les mécanismes d’ionisation se ressemblent beaucoup, mais changent quant à leur utilisation. Le concept d’impact électronique est utilisé directement pour certaine sources tandis que, dans d’autres il sert seulement à initier les réactions. C’est au chercheur à bien déterminer la méthode qui sied le mieux à l’échantillon à
analyser. On verra qu’à la pression atmosphérique, on s’inspire de différentes méthodes d’ionisation pour construire des sources plus efficaces.
2.3 Les sources et méthodes d’ionisation à la pression atmosphérique
La particularité des sources d’ionisation à la pression atmosphérique est qu’on doit introduire un agent extérieur (l’échantillon) dans une enceinte sous vide (le spectromètre de masse). Le développement de ces méthodes d’ionisation commença lorsqu’on a voulu coupler le spectromètre de masse avec un chromatographe en phase liquide ou gazeuse. Les avantages découlant de ces nouvelles méthodes permirent de développer de multiples sources pouvant ioniser l’échantillon présent sous différentes formes. Un de ces avantages est la douceur d’ionisation qui permet d’analyser des composés biologiques de grande masse (jusqu’à des millions de Daltons). Ceci est devenu aujourd’hui un très grand champ d’intérêt, où l’on s’applique à analyser et détecter des protéines, des peptides, des lipides, etc. Un autre avantage est que l’ionisation à la pression atmosphérique permet de réduire l’énergie excédentaire des ions rapidement ce qui empêche ceux-ci de se dénaturer. Enfin, on peut maintenant introduire des échantillons en phase solide dans une source.
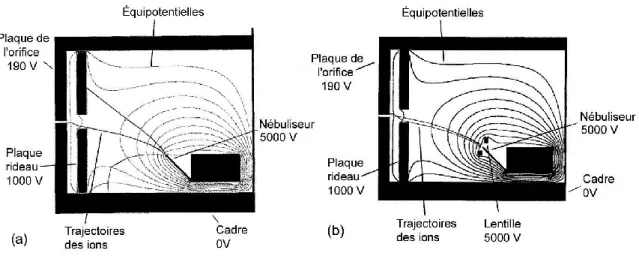
Comme le spectromètre de masse opère dans un vide minimum de 10-5 Torr, la source ionique à la pression atmosphérique est devenue un module extérieur à l’appareil. Cependant, l’introduction d’ions nécessite de les faire passer de 760 Torr à la pression du spectromètre de masse. On inventa l’interface à la pression atmosphérique (API) qui consiste en une succession d’étapes de pompage (figure 2.2). Chaque étape a une pression inférieure à la précédente et comporte des éléments focalisant le faisceau d’ions. Le jet moléculaire supersonique prend place dans le premier étage habituellement. L’écorceur (forme conique dans la figure 2.2) vient accueillir les analytes se retrouvant au centre du jet.
23
Figure 2.2: Interface à la pression atmosphérique 16
L’électro-nébulisation (ESI) ainsi que l’ionisation chimique à la pression atmosphérique (APCI) sont utilisées pour ioniser un jet liquide provenant d’un chromatographe en phase liquide ou d’un autre système. Les sources AP-MALDI (Atmosheric Pressure Matrix Assited Laser Desorption Ionization), DESI (Desorption Electrospray Ionization) et LDTD ionisent, quant à elles, les analytes provenant de solutions en phase solide ayant été désorbées par différentes techniques. L’ionisation se fait généralement par des réactions ions-molécules et transfert de proton tel que décrit pour l’ionisation chimique. Pour certaines, la grande quantité de solvant utilisée pour traiter l’échantillon nécessite un procédé de désolvatation avant qu’elle entre dans le spectromètre de masse. On présente ici trois sources qui sont différentes au niveau de l’ionisation, de la désolvatation ainsi que de la désorbtion des analytes. Ces instruments utilisent les différentes méthodes d’ionisation classiques vues à la section précédente (2.2).
2.3.1 L’électro-nébulisation
L’électro-nébulisation (Electrospray ionization ESI) est une technique qui permet de coupler le spectromètre de masse avec un chromatographe en phase liquide. Un capillaire amène la phase liquide au centre d’une électrode tubique en acier inoxydable de petit rayon ayant un potentiel de plusieurs kilovolts. Le champ électrique produit une accumulation de charges à la surface du liquide. Cette surface se déformera pour devenir sous l’effet du champ très pointue (aussi appelée cône de Taylor). La rupture du cône produit des gouttelettes chargées qui seront nébulisées dans l’espace séparant le capillaire du spectromètre de masse (figure 2.3). À ce stade la phase liquide a été évaporée, mais le solvant présent dans la solution initiale est toujours mélangé avec les analytes. Ainsi, les goutelettes sont amenées dans une chambre de désolvatation qui peut prendre la forme d’un capillaire ou d’une enceinte chauffée. On utilise aussi un gaz sec (N2) et chaud pour faire évaporer le solvant. Pendant que le solvant s’évapore, la densité
de charges de même polarité augmente. Lorsque la force de répulsion de ces charges devient supérieure à la tension de surface de la gouttelette, des ions sont libérés. Puis, les ions entrent dans le spectromètre de masse par l’interface à la pression atmosphérique. La distance séparant le capillaire de l’entrée du spectromètre de masse dépend de la longueur de cette chambre qui peut varier d’une source à une autre.
Le capillaire peut être positionné en face de l’entrée du spectromètre de masse, mais le flux étant directement dirigé vers celle-ci, on favorise l’introduction de solvant et de molécules neutres dans l’appareil. Pour diminuer ce phénomène, certaines sources placent le capillaire de façon à ce que le jet soit perpendiculaire à l’entrée. Les configurations utilisées favorisent la dispersion des ions dans l’espace les séparant de l’entrée de l’appareil. Ultimement, seulement une partie des analytes peut être mesurée.
25
Figure 2.3: Principe de fonctionnement d’une source à électro-nébulisation 17
L’ionisation à proprement dite des analytes se produit lorsque le solvant est complètement évaporé de l’échantillon pour les grosses molécules ou bien lorsque les analytes ionisés se désorbent des gouttelettes pour les petites molécules. On peut considérer que l’ionisation se fait par transfert de charges entre molécules. On parle donc d’ionisation chimique si on considère que le solvant transporte majoritairement la charge et que, lors de l’évaporation, il laisse sa charge à l’analyte. L’ionisation ne s’apparente pas totalement à un des types décrits classiquement, mais est un résultat de plusieurs de ces phénomènes.
Comme la nébulisation rapide de la solution empêche celle-ci d’atteindre un équilibre, les analytes se retrouvent ultimement avec un surplus de charges (+/-). Les charges présentes dans la phase gazeuse proviennent des électrons fournis par le capillaire (oxydation) pour le mode positif et le contraire (réduction) en mode négatif. Comme l’attachement d’électrons est limité par plusieurs paramètres, le nombre de charges disponibles pour ioniser est équivalent au courant produit par les électrons. Typiquement, les sources d’électro-nébulisation produisent des courants de 1 μA. La pression atmosphérique et le fait que l’échantillon ne soit pas chauffé ou très peu, font en sorte que les ions sont
stables et ne se fragmentent pas. Cette méthode est excellente pour analyser des molécules non volatiles ou difficilement évaporables et des molécules thermolabiles. L’évaporation rapide du solvant favorise la production d’ions multichargés ce qui permet de détecter des molécules ayant des poids élevés avec des spectromètres de masse mesurant des rapports m/z limités.
2.3.2 AP-MALDI
La technologie MALDI se démarque des autres méthodes discutées précédemment, par le fait que la solution est introduite dans la source en phase solide. La source MALDI (figure 2.4) a été conçue initialement pour être utilisée sous vide, mais son évolution permet aujourd’hui de l’utiliser à la pression atmosphérique. Les mécanismes d’ionisation sont très semblables pour les deux sources. L’échantillon est mélangé dans une solution comprenant des solvants ainsi que des molécules organiques capables d’absorber une impulsion lumineuse laser. L’échantillon est déposé sur un support, puis séché pour évaporer le solvant. Le support est par la suite introduit dans la source où un faisceau laser est dirigé sur la solution solide ce qui permet de la désorber. Cette désorption est due à l’absorption de l’énergie des photons par les molécules organiques. Cette énergie intense et de courte durée ne permet pas aux molécules de l’évacuer normalement. Celles-ci acquièrent alors de l’énergie cinétique et désorbent. L’efficacité de cette technique réside dans la nature de la matrice. Elle doit être en mesure d’absorber la longueur d’onde du laser utilisé ainsi que, par la suite, se dissocier des analytes. On peut ioniser des molécules ayant des masses élevées (> 100 000 Da). Pour ne pas dégrader les analytes, la quantité de molécules organiques doit être très supérieure à ceux-ci.
Les mécanismes d’ionisation sont très variés en MALDI et ne sont pas très bien quantifiés. L’ionisation se produit entre autres par photo-ionisation, réactions ions-molécules et transfert de proton. Toutefois, l’ionisation y est relativement bonne, car le rendement
27 d’une telle source est excellent. Cet appareil est disponible commercialement. En AP-MALDI, les ions n’ont pas tendance à se fragmenter, contrairement à ce qui se produit dans le vide, mais ils tendent à faire des agglomérations avec la matrice. Cet effet peut rendre les spectres très difficiles à interpréter.
Figure 2.4: Schéma de fonctionnement d’une source MALDI 73.
Le support à échantillon est à un potentiel électrique pour permettre aux ions de dériver vers l’entrée du spectromètre de masse. Pendant cette dérive, les ions ont tendance à se disperser comme pour l’électro-nébulisation.