Étude de la microflore des fromages du terroir
québécois par métabarcoding
Mémoire
Annick Raymond-Fleury
Maîtrise en sciences et technologie des aliments - avec mémoire
Maître ès sciences (M. Sc.)
Étude de la microflore des fromages du terroir
québécois par métabarcoding
Mémoire
Annick Raymond-Fleury
Sous la direction de :
Résumé
L’industrie des fromages de spécialité occupe une place très importante au Québec. Sa production annuelle représente plus de 60 000 tonnes de fromages. Bien que les produits québécois se démarquent par leur haute qualité, ils manquent parfois de constances au niveau de leurs propriétés sensorielles (goût, odeur, texture, couleur). Ces variations peuvent être expliquées en partie par un déséquilibre de la microflore fromagère (bactéries et mycètes). Bien que la microflore joue un rôle majeur dans le développement des fromages, très peu d’information est disponible sur sa composition (présence et abondance relative des espèces) pour les fromages du terroir québécois.
Le but de ce projet de recherche est de développer une méthode de métagénomique ciblée par séquençage massif d’amplicons (metabarcoding) pour faire la caractérisation complète de la microflore de la surface (croûte) et du cœur (pâte) de ces fromages. Le métabarcoding est une méthode d’identification qui utilise une courte séquence d’ADN représentative du génome entier. Les résultats obtenus lors de ce projet montrent que la région V3-V4 de l’ADNr 16S, pour les bactéries, et la région ITS2 de l’espaceur de transcription interne, pour les mycètes, sont les régions qui permettent de dépeindre le portrait le plus fidèle des écosystèmes fromagers.
La microflore de 32 fromages du terroir québécois a été caractérisée. Les régions cibles utilisées recensent les genres dominants associés aux écosystèmes fromagers et permettent aussi la détection spécifique de certains genres moins fréquents. Le nombre de genres dominants identifiés est de 20 pour la région V3-V4, de 22 pour la région V6-V8, de 12 pour la région ITS1 et de 13 pour la région ITS2. Il a également été possible de comparer la communauté microbienne de 15 fromages pour deux années de production (2015 et 2018, 30 fromages au total) afin d’observer la variation de la microflore en fonction du temps. Cette étude a permis d’observer que plus de la moitié des écosystèmes étudiés se révèle être constante. Ces nouvelles connaissances permettent de mieux décrire la typicité des fromages québécois et de proposer des leviers technologiques aux artisans pour produire des fromages de haute qualité de façon plus constante.
Abstract
The speciality cheese industry plays an important role in the province of Quebec, with a production of over 60 000 tonnes of cheeses. Although these products distinguish themselves with their high quality, a lack of constancy is sometime experienced regarding their sensory properties (taste, smell, texture, color). These variations can be explained, partly, by a microflora disequilibrium (bacteria and fungi). Even though microflora plays an important role in cheese ripening, very little information is available about his composition (presence and relative abundance of different species) for Quebec’s terroir cheeses.
This research project aims to develop a targeted metagenomic by massive amplicon sequencing (metabarcoding) to characterize the microflora of cheese rind and cheese core. Metabarcoding is a profiling method using short sequences representative of the entire genome. In this project, the V3-V4 region of the 16S rDNA and the ITS2 region of the rDNA ITS region were identified as the most precise molecular markers for the profiling of respectively bacterial and fungal cheese ecosystems.
The microflora of 32 cheeses of the Quebec terroir has been characterized. The target regions used identified the dominant genera associated with cheese ecosystems and also allow the specific detection of some less frequent genera. The number of dominant genus assigned is 20 for the V3-V4 region, 22 for the V6-V8 region, 12 for the ITS1 region and 13 for the ITS2 region. It was also possible to compare the microbial community of fifteen cheeses for two different years of production (2015 and 2018) in order to observe the variation of the microflora over time. This study shows that over half of the ecosystems analyzed are stable. This new knowledge allows a better understanding of Quebec’s terroir cheese typicity and offers new information to cheesemakers on the way to produce high quality cheeses more consistently.
Table des matières
Résumé ... ii
Abstract ... iii
Table des matières ... iv
Liste des figures ... vii
Liste des tableaux ... x
Liste des acronymes ... xi
Remerciements ... xii
Avant-propos ... xiii
Introduction ... 1
Chapitre 1 – Revue de littérature ... 3
1.1 Fabrication fromagère ... 3
1.2 La microflore des fromages ... 4
1.2.1 La microflore bactérienne primaire des fromages ... 4
1.2.2 La microflore bactérienne secondaire et la microflore d’affinage des fromages ... 6
1.2.3 La microflore fongique utile des fromages ... 8
1.2.4 La microflore dépendante du type de technologie fromagère ... 10
1.2.5 La microflore et la production d’arômes ... 12
1.3 La typicité fromagère et la notion de terroir ... 12
1.4 Les méthodes d’identification du microbiote fromager ... 13
1.4.1 Les analyses phénotypiques de microorganismes isolés ... 13
1.4.2 Les analyses moléculaires non ciblées ... 14
1.4.3 Le séquençage du génome complet d’un organisme... 17
1.4.4 Le barcoding ... 18
1.4.5 Le métabarcoding ... 18
1.5 La notion d’unité taxinomique opérationnelle ... 21
1.6 La biodiversité ... 22
1.6.1 Indice de richesse ... 22
1.6.2 Indice d’alpha-diversité ... 22
1.6.3 Indice de bêta-diversité ... 23
1.7 L’analyse bio-informatique ... 23
1.7.2 L’appariement des séquences ... 26
1.7.3 L’élimination des séquences chimériques ... 26
1.7.4 Le regroupement des séquences en UTOs ... 27
1.7.5 L’assignation taxinomique ... 29
1.8 Les applications du métabarcoding ... 29
Chapitre 2 – Problématique, hypothèse et objectifs ... 31
2.1 Problématique ... 31
2.2 Hypothèse de recherche ... 32
2.3 Objectifs de recherche ... 32
Chapitre 3 – Persistence of Quebec’s terroir cheese microflora using metabarcoding ... 33
3.1 Résumé ... 33
3.2 Abstract ... 34
3.3 Importance ... 34
3.4 Introduction ... 35
3.5 Results ... 36
3.5.1 Sequencing Data and Taxonomic Assignation... 36
3.5.2 Choice of the Targets for Cheese Microflora Metabarcoding ... 38
3.5.3 Microbial Landscape of Quebec’s Terroir Cheeses ... 41
3.5.4 The Consistency of Cheese Ecosystems ... 46
3.6 Discussion ... 47
3.6.1 Selecting the Genomic Target for Cheese Metabarcoding ... 47
3.6.2 Assessing the Stability of Quebec’s Terroir Cheese Microflora Over Years ... 48
3.7 Materials and Methods ... 51
3.7.1 Cheese sampling ... 51
3.7.2 DNA Extraction, Library Construction and Sequencing ... 51
3.7.3 Accession Number ... 52
3.7.4 Bioinformatic Analysis ... 52
3.7.5 Metrics and Figures... 53
3.7.6 Acknowledgement ... 53
Chapitre 4 – Discussion générale ... 54
4.1 Retour sur les résultats ... 54
4.2 Limites de l’étude ... 55
Conclusion générale et perspective ... 56 Bibliographie ... 57
Liste des figures
Figure 1 Aspect visuel de fromages de spécialité. (A) Fromage à croûte lavée, de couleur orangée. (B) Fromage à croûte mixte, dont l’apparence est intermédiaire. (C) Fromage à croûte fleurie, dont la surface est blanche et duveteuse. ……….………..10
Figure 2 Schématisation du séquençage de Sanger. (À gauche) Représentation de la migration des fragments générés à la suite de la réaction de polymérisation sur gel d’électrophorèse capillaire et détection de la couleur associée à chaque ddNTP. (À droite) Représentation du fragment synthétisé par polymérisation à la suite de l’arrêt lors de l’incorporation d’un ddNTP à l’extrémité 3’ et déduction de la séquence d’ADN résultante...15
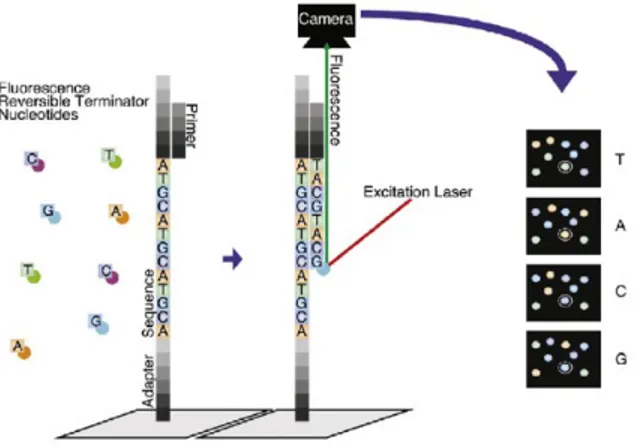
Figure 3 Technologie de séquençage à haut débit d’Illumina. (À gauche) Simple brin d’ADN fixé à la plaque par un adaptateur, auquel une amorce est hybridée afin de débuter l’élongation, en présence de nucléotides terminateurs réversibles fluorescents. (Au centre) À la suite de l’ajout du nucléotide, un signal lumineux est émis et la fluorescence est interprétée par une caméra. (À droite) Cette opération se réalise en parallèle à plusieurs endroits sur la plaque. L’ensemble des signaux est compilé afin de connaître la séquence de chaque fragment……….………...18
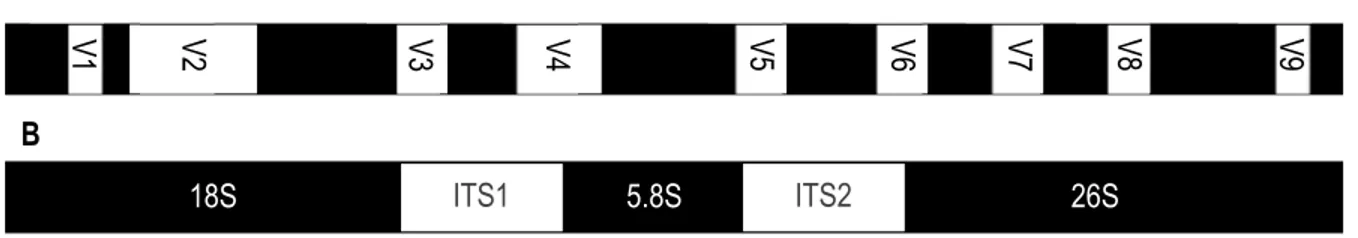
Figure 4 Représentation des cibles de l’ADNr bactérien (A) et fongique (B) utilisées en métabarcoding. (A) L’ADNr 16S bactérien comporte neuf régions hypervariables, numéroté de V1 à V9. (B) L’ADNr fongique présente deux espaceurs de transcription interne, ITS1 et ITS2……….………..……21
Figure 5 Importance de la richesse (en haut) et de l’équitabilité (en bas) pour la définition de la diversité. (En haut) Une communauté de 7 espèces semble plus diverse qu’une communauté de 2 espèces. (En bas) À richesse égale, une communauté moins équitable (à gauche) semble moins diverse. (À gauche) Une communauté moins riche (en haut) peut sembler plus diverse si elle est plus équitable. (À droite) Idem pour la communauté du bas...……….……….…22
Figure 6 Principe de la fenêtre glissante de Trimmomatic. L’analyse de la qualité est effectuée une première fois, puis si la valeur est au-dessus du seuil fixé, l’analyse se poursuit avec un décalage d’un nucléotide en aval. L’analyse se poursuit jusqu’à ce que la qualité dans l’étendue de la fenêtre soit inférieure au seuil fixé…...….25
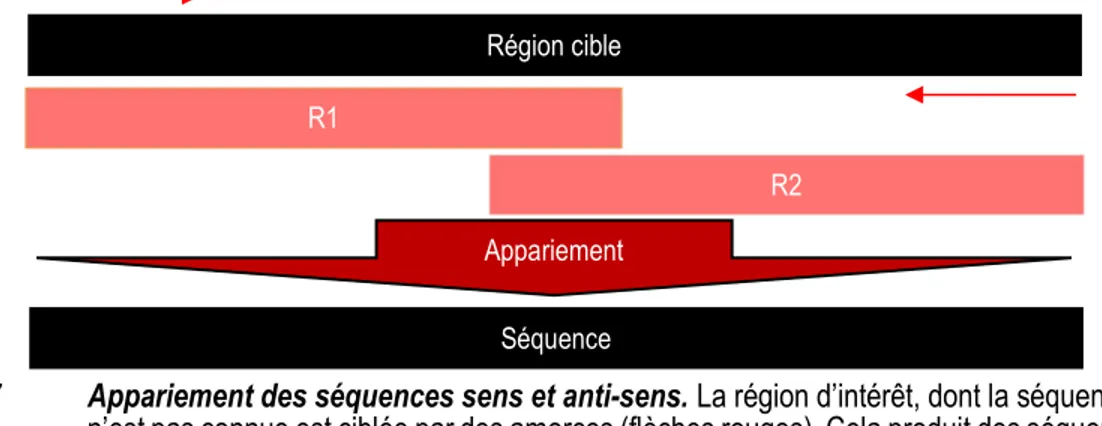
Figure 7 Appariement des séquences sens et anti-sens. La région d’intérêt, dont la séquence exacte n’est pas connue est ciblée par des amorces (flèches rouges). Cela produit des séquences sens (R1) et anti-sens (R2) que l’on peut ensuite apparier afin de connaître la séquence de la région d’intérêt………...…...26
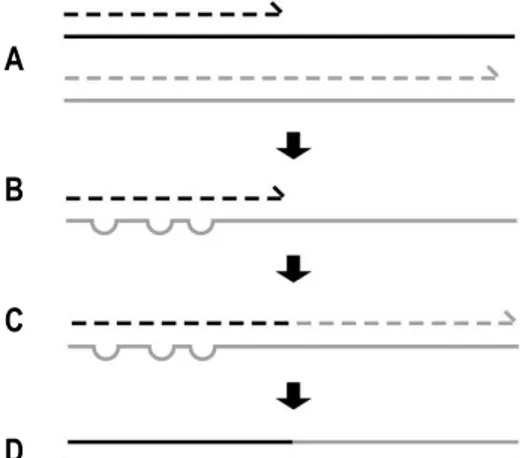
Figure 8 Formation d’une chimère lors de l’amplification PCR. (A) Brin d’ADN dont l’extension a été interrompue prématurément lors d’un cycle de PCR (en noir). (B) Ce court brin peut s’hybrider à un brin d’ADN d’une autre origine et partiellement homologue (en gris), permettant quelques mésappariements (hétéroduplex). (C) La courte séquence sert d’amorce et il y a élongation du court brin. (D) Cette séquence chimérique (grise et noire) sera amplifiée dans les cycles suivants de PCR, mais ne correspond pas à une molécule d’ADN retrouvée dans l’écosystème…..……….………..……...27
Figure 9 Schématisation de l'approche par seuil d’identité et par Swarm. (A) Visualisation de la formation des UTOs, selon un seuil d’identité (t) (généralement 97 %). Le choix de la première séquence (en rouge) a un impact sur la formation des UTOs, car des amplicons étroitement liés peuvent être placés dans différents UTOs. (B) Swarm fait des regroupements de façon itérative, en utilisant un petit seuil de regroupement (d), choisi par l'utilisateur, permettant aux UTOs de regrouper toutes les séquences semblables……….………..28
Figure 10 Compared distribution of the most abundant eukaryotic orders in cheese ecosystems………..39
Figure 11 Compared distribution of the most abundant bacteria in cheese ecosystems………....………..40
Figure 12 Compared distribution of the most abundant eukaryotic genera in cheese ecosystems………..………42
Figure 13 Compared distribution of the most abundant bacterial genera in cheese ecosystems………...…………43
Liste des tableaux
Tableau 1 Odeur des composés volatils issus du métabolisme de la microflore
fromagère……….……12
Tableau 2 Comparaison des plateformes de NGS.……….………...17
Tableau 3 Régions cibles de l’ADNr utilisées pour étudier les écosystèmes fromagers...…20
Tableau 4 Caractéristiques des outils pour le nettoyage des séquences Illumina...………..25
Table 5 Information on cheese sampled………..………..…..37
Table 6 Sequencing data of the fungal (ITS1 and ITS2) and bacterial (V3-V4 and V6-V8) ecosystems………..37
Table 7 Richness and diversity of cheese microflora………...44
Table 8 Constancy of cheese microbiota according to the Bray-Curtis index……….46
Liste des acronymes
ADNr : acide désoxyribonucléique ribosomique;
AEM : Applied and Environmental Microbiology;
dNTP : désoxyribonucléotides;
ddNTP : didésoxyribonucléotides;
FAO : Organisation des Nations Unies pour l’alimentation et l’agriculture (en anglais, food and agriculture organization of the United Nations);
HTS : séquençage à haut débit (en anglais, high-throughput sequencing);
IBIS : Institut de biologie intégrative et des systèmes;
ITS : internal transcribed spacer;
N.A. : Not available;
NGS : séquençage de nouvelle génération (en anglais, next-generation sequencing);
OTU : operational taxonomic unit;
pb : paire de bases;
PCR : réaction en chaîne de la polymérase (en anglais, polymerase chain reaction);
PEAR : Paired-End reAd mergeR;
qPCR : PCR quantitative;
T-RFLP: polymorphisme de longueur des fragments de restriction terminaux (en anglais, terminal restriction fragment-length polymorphism);
Remerciements
Je tiens à remercier toute l’équipe du laboratoire de mycologie alimentaire, et tout particulièrement mon directeur de recherche, Steve Labrie, qui a cru en mon potentiel. Merci Steve de m’avoir accompagné, conseillé et encouragé tout au long de mon parcours. J’ai toujours senti que ta porte m’était grande ouverte, ce que j’ai grandement apprécié. Je remercie Marie-Hélène Lessard pour sa disponibilité et son aide précieuse. Je remercie également tous les membres de l’équipe qui ont contribué de près ou de loin à la réussite de ces travaux : Catherine Viel, Andréanne, Stéphanie, Vincent, Gabrielle, Laura, Virginie et Momo. Je remercie aussi tous les étudiants en Sciences des aliments qui ont croisé ma route et qui ont contribué à ma culture scientifique.
Un grand merci à mon co-directeur Daniel St-Gelais pour ses discussions constructives et ses connaissances complémentaires qui ont su m’éclairer aux moments opportuns.
Je remercie tous les partenaires financiers qui ont permis la réalisation de ces travaux et je tiens à souligner la participation des nombreux fromagers qui ont accepté de collaborer à cette étude. Sans vous, rien de tout cela n’aurait été possible. Merci de votre confiance et de votre générosité!
Pour conclure, je ne saurais exprimer toute ma gratitude envers ces deux personnes qui m’ont permis de surmonter ce défi et de parvenir à l’aboutissement de ce périple : Julien Chamberland et Steven Gallant. Merci Julien de m’avoir partagé ta bonne humeur contagieuse, ton énergie positive et d’avoir été une source intarissable de conseils. Tu auras su me guider alors que j’en avais le plus besoin. Merci Steven d’avoir été le support moral dont j’ai eu si souvent besoin. Merci de m’avoir accompagné à travers cette tumultueuse aventure, et d’accepter d’en partager une encore plus grande et merveilleuse très bientôt.
Avant-propos
Ce mémoire est divisé en cinq sections. La première section est l’introduction, qui met en contexte la problématique de ce projet de maîtrise. La deuxième section est une revue de la littérature résumant les connaissances actuelles sur la microflore des fromages et les méthodes disponibles afin de la caractériser. Cette section se termine par la présentation de la problématique de recherche, l’hypothèse et les objectifs de ce projet de maîtrise.
La troisième section est écrite sous forme d’article scientifique, en anglais, et présente tous les résultats obtenus dans le cadre de ce projet. L’article sera soumis pour publication dans la revue scientifique Applied and Environmental Microbiology (AEM). L’auteure de ce mémoire a réalisé la plus grande partie du travail expérimental, dont l’élaboration du protocole bio-informatique, de l’analyse des résultats et de la rédaction de l’article scientifique, dont elle est la première auteure. Le séquençage a été réalisé par la plateforme d’analyse génomique de l’institut de biologie intégrative et des systèmes (IBIS) de l’Université Laval. Le Dr Julien Chamberland a participé activement à l’élaboration initiale du protocole d’analyse bio-informatique et à l’écriture de l’article scientifique, lors de ses études sous la direction du Dr Yves Pouliot. Les Drs Julien Chamberland, Éric Dugat-Bony, Sylvie Turgeon et Daniel St-Gelais ont participé à la révision de l’article scientifique. La Dre Marie-Hélène Lessard a cosupervisé le travail, a contribué à la correction du mémoire et a participé à l’écriture de l’article scientifique. Le Dr Steve Labrie a obtenu les fonds de recherche, a supervisé le travail, a contribué à la révision de l’article et a participé activement à l’écriture du mémoire. La majorité des résultats de cet article ont été présentés sous forme d’affiche intitulée « Étude de la typicité des fromages québécois par métabarcoding » lors du Forum technologique Novalait à Saint-Hyacinthe (Québec, Canada) en 2018.
La quatrième section offre une discussion générale sur les travaux présentés dans le mémoire, afin de révéler les principaux résultats de ce projet de maîtrise et de mettre de l’avant l’importance de ces découvertes, ainsi que les limitations de cette étude. La cinquième et dernière section est une conclusion générale et présente les perspectives du projet.
Introduction
L’industrie fromagère occupe une place très importante au Canada. Au pays, 510 250 tonnes (t) de fromages ont été produites en 2018. Près du tiers (164 403 t – 32 %) de la production est associée à la catégorie des fromages de spécialité [1]. Le Québec joue un rôle de premier plan dans cette transformation, puisqu’on y produit annuellement près du tiers des fromages de spécialité au pays.
On retrouve 535 fromages fermiers et artisanaux au Québec, dont 315 sont fabriqués à partir de lait de vache [2]. Ces fromages rencontrent souvent les plus hauts standards de qualité et remportent des prix prestigieux [3], mais ils manquent parfois de constance au niveau de leurs caractéristiques sensorielles. Cette inconstance provient en grande partie du lait utilisé pour leur fabrication, de sa composition microbiologique et de ses propriétés technologiques. Bien que la microflore joue un rôle majeur dans le développement des propriétés sensorielles des fromages, très peu d’information est disponible sur sa composition (présence et abondance relative) pour les fromages du terroir québécois. Le recensement des écosystèmes fromagers permet d’identifier les microorganismes jouant un rôle d’intérêt dans l’affinage des fromages.
Les fromages de spécialité développent une microflore riche et diversifiée, tant en surface que dans la pâte du fromage. La matière première, le lait, est inoculée par un mélange complexe de ferments d’acidification et d’affinage qui contribuent aux caractéristiques sensorielles des fromages [4]. Cependant, la microflore secondaire provenant majoritairement de l’environnement va également s’établir au fil du temps pouvant même devenir majoritaire dans certaines variétés. Cet écosystème dynamique complexe évolue d’une façon difficile à prévoir et à contrôler dans le produit final. Ceci est principalement dû à la méconnaissance de toutes les espèces microbiennes qui se développent et des paramètres de fabrication qui les affectent. La compréhension de l’écosystème fromager est nécessaire afin de contribuer au contrôle de l’affinage des fromages [5-7].
Pendant plusieurs dizaines d’années, des méthodes de microbiologie traditionnelle ont été utilisées afin de caractériser l’écosystème fromager [8]. Cependant, les méthodes de culture ne permettent pas de détailler l’ensemble de l’écosystème fromager puisque plusieurs espèces de microorganismes peuvent être difficilement cultivables ou être dans un état de
survie nommé « viable, mais non-cultivable » [9, 10]. Également, les méthodes de culture ont souvent tendance à favoriser la détection des microorganismes majoritaires au détriment des espèces minoritaires. Dans les dernières années, de nouvelles méthodes comme la métagénomique et le métabarcoding ont fait leur apparition permettant d’évaluer les espèces composant la microflore globale et leur abondance relative dans un écosystème. Ceci a permis de détecter des microorganismes dont la présence était insoupçonnée jusqu’alors [7, 11]. Ces études ont été réalisées pour des fromages de plusieurs origines à travers le monde. Celles-ci montrent que le portrait de la composition microbienne et la complexité de la microflore varient d’une variété de fromages à l’autre [7, 12, 13]. Pour les fromages à croûte lavée, plus d’une centaine de microorganismes ont pu être identifiés alors que moins d’une dizaine d’espèces sont inoculées lors du procédé de fabrication/affinage [6, 7, 14]. Il est aussi possible de suivre les activités métaboliques de plusieurs espèces lors de l’affinage des fromages à l’aide de la transcriptomique [12, 13].
Les fromagers d’ici sont à la recherche de connaissances et de moyens permettant de réduire l’impact des inconstances de qualité. Une meilleure compréhension de la microflore fromagère permettrait de fournir des leviers technologiques aux artisans afin de mieux contrôler l’affinage de leurs fromages et d’identifier les espèces typiques à leur production [15]. En caractérisant les écosystèmes, il serait éventuellement possible de modifier les cultures d’appoints inoculées au lait lors de la préparation des fromages, permettant ainsi une « standardisation » de la microflore lors de la production fromagère.
Au Québec, la caractérisation fongique et chimique des laits servant à la fabrication de fromages du terroir québécois a été réalisée [16] et trois levures d’intérêt dans l’affinage des fromages du terroir ont été étudiées pour leur capacité fromagère [17]. Cependant, à ce jour, aucune étude utilisant des méthodes issues de la génomique n’a été utilisée afin de décrire l’écosystème des fromages d’ici. Ce projet de recensement par l’utilisation du métabarcoding pour caractériser les fromages de plusieurs fromageries du terroir est une première et vise une meilleure compréhension de la microflore des fromages et de la typicité des fromages québécois.
Chapitre 1 – Revue de littérature
1.1 Fabrication fromagère
Le fromage est un produit alimentaire élaboré à base de lait. L’origine animale du lait peut être aussi diverse qu’il existe de mammifères, mais les plus communs sont les fromages à base de lait de vache, de chèvre, de brebis et de bufflonne [18]. La composition du lait (teneur en protéines et en matières grasses, par exemple) et sa microflore varient en fonction des mammifères, ce qui peut générer différents profils de propriétés sensorielles [18]. Le lait utilisé en production fromagère peut être soumis, ou non (lait cru), à un traitement thermique (lait pasteurisé ou thermisé), ce qui aura une incidence sur la charge microbiologique initiale du lait [19-21].
La fabrication du fromage débute par l’ajout de chlorure de calcium (CaCl2), de ferments
lactiques et d’enzymes, telles que la présure, ce qui entraîne la coagulation des micelles de caséines (coagulation lactique ou enzymatique). Pour les fromages affinés en surface, l’ajout des ferments d’affinage directement dans le lait permet leur meilleure répartition dans l’ensemble du caillé, mais ceux-ci peuvent également être déposés sur les fromages par vaporisation et lors des soins de croûte (lavage) dans le cas des affinages en surface [22]. L’étape de coagulation, influencée par la concentration d’enzymes, la quantité de calcium, le pH et le temps d’entreposage [4], provoque la formation d’un gel. Lorsqu’il est découpé et brassé, le gel génère deux phases, soit le caillé et le lactosérum, qui sont séparées lors de l’égouttage. Le caillé récupéré est alors mis en moule et retourné pour éliminer le lactosérum. L’égouttage peut se faire lentement, par gravité, pour les fromages à pâte molle, ou de façon accélérée par pressage et parfois par cuisson, pour les fromages à pâte semi-ferme et semi-ferme [4]. Généralement, une fois l’égouttage terminé, les fromages sont démoulés puis salés par immersion en saumure ou par salage à sec [4]. Pour certaines variétés, ces dernières étapes peuvent avoir lieu dans un ordre différent. Les fromages frais sont alors prêts à la consommation.
La majorité des variétés de fromages démoulés passent par une étape finale d’affinage dans des conditions contrôlées d’humidité et de température. Pendant cette période, qui peut être de quelques jours (ex. Port-Salut) à plusieurs mois (ex. Cheddar), les ferments d’affinage et la microflore secondaire se développent, ce qui confère aux fromages leurs caractéristiques
sensorielles typiques [4]. Les fromages à croûte fleurie, comme le Camembert et le Brie, se recouvrent de levures et de moisissures qui leur confèrent une apparence duveteuse, tandis que les fromages à croûte lavée (généralement frottés à quelques reprises avec une solution saline contenant des ferments d’affinage) se recouvrent d’une croûte humide et souvent orangée composée principalement de bactéries (ex. Brevibacterium sp., Glutamicibacter sp.) et de quelques espèces de levures (ex. Geotrichum sp., Debaryomyces sp., Kluyveromyces sp.) [23].
Ces différentes étapes sont autant de leviers technologiques qui permettent aux maîtres-fromagers d’obtenir différentes variétés de fromages possédant chacune des propriétés texturales et organoleptiques qui leur sont propres. Les fromages peuvent être classés selon les technologies utilisées, telles que le type de coagulation (caillé de type lactique, présure ou mixte), le taux d’humidité (pâte molle, semi-ferme ou ferme) ou le type de croûte (fleurie, lavée ou mixte), pour en nommer que quelques-unes [24].
1.2 La microflore des fromages
La microflore des fromages est composée d’un grand nombre de microorganismes qui peut atteindre entre 2 x 109 et 3 x 109 UFC/g de fromage [4]. Il s’agit d’un ensemble de bactéries,
de levures et de moisissures qui proviennent à la fois de l’environnement et d’inoculations volontaires (ferments et cultures d’affinage). La distribution de la microflore est très hétérogène au sein d’un même fromage, la composition microbiologique (quantité, diversité) varie énormément entre la surface (croûte) et le cœur de la meule (pâte) [25].
1.2.1 La microflore bactérienne primaire des fromages
Lors de la fabrication fromagère, les ferments lactiques et d’affinage sont ajoutés directement dans le lait avant d’être mélangés. Ce sont les microorganismes qui ont souvent un rôle dans l’acidification du lait et/ou la génération de composés aromatiques. Après le démoulage, le fromage prend sa forme définitive, et le cœur de la meule n’est plus en contact avec l’environnement extérieur. Les microorganismes retrouvés dans la pâte et à la surface de fromages frais ou peu affinés (fromage en début d’affinage, Mozzarella) correspondent aux ferments ajoutés lors de la production [4, 26-28]. La microflore bactérienne se compose généralement des ferments lactiques, appartenant aux genres bactériens Lactococcus, Streptococcus, Leuconostoc et Lactobacillus [29, 30]. Les
bactéries lactiques, qui contribuent à l’acidification du caillé, favorisent l’implantation d’une microflore acidophile, soit les levures et les moisissures filamenteuses [31, 32].
Le genre Lactococcus est couramment utilisé comme ferment lactique d’acidification. En début d’affinage, les lactocoques sont dominants, avec plus de 109 UFC/g de fromage [4].
Les sous-espèces Lactococcus lactis subsp. lactis et L. lactis subsp. cremoris sont les plus utilisées [33]. Ces bactéries ont un rôle majeur dans la production des fromages puisqu’elles métabolisent le lactose en acide lactique, ce qui acidifie le milieu (fermentation lactique) et permet la coagulation des caséines. De plus, les lactocoques ont une importante activité protéolytique et leur principale enzyme, la protéase PrtP, est une protéine kinase qui a la capacité d’hydrolyser les caséines [34]. Les lactocoques diminuent le potentiel redox en début d’affinage et contribuent aux flaveurs des fromages de type Cheddar, Gouda ou Saint-Paulin par la production de métabolites secondaires (acétaldéhyde, éthanol, acide acétique) [35]. Cependant, certaines souches de Lactococcus sp. ont une activité indésirable lors de l’affinage en contribuant à l’amertume des fromages [34].
Streptococcus thermophilus est une espèce thermophile (croissance optimale entre 40-45 C) souvent utilisée en combinaison avec d’autres bactéries lactiques pour la fabrication de fromages à pâte pressée (ex. Emmental, Gruyère, Gorgonzola) [36] et à pâte molle stabilisée [4]. En effet, cette espèce peut être utilisée pour raffermir la pâte de fromages à croûte fleurie, pour produire des fromages dits à pâte stabilisée [37]. Son rôle au cours de l’affinage est semblable à celui des lactocoques, mais leur production en acétaldéhyde est beaucoup plus importante et confère des arômes de fraicheur recherchés chez les variétés des fromages concernées [4].
Leuconostoc mesenteroïdes est surtout utilisé pour sa production de diacétyle et d’acétoïne, des composés qui confèrent un goût de beurre aux fromages [38]. Ces bactéries hétérofermentaires produisent également de l’éthanol et de l’acide acétique qui contribuent aux arômes des fromages et du dioxyde de carbone (CO2) qui mène à l’apparition
d’ouvertures dans la pâte [4, 39, 40]. La charge microbienne de Leuconostoc est très variable selon les variétés de fromages, allant de 103 à 109 UFC/g, et ils sont habituellement
Les Lactobacillus sp. se retrouvent en grand nombre (107-108 UFC/g de fromage) dans
les pâtes pressées cuites. Les lactobacilles sont le plus souvent utilisés comme culture d’appoint [42]. Ils ont un rôle mineur dans l’acidification du caillé, mais interviennent pendant l’affinage des fromages par leurs activités protéolytiques, peptidasiques, lipasiques et estérasiques [43]. Leur activité protéolytique est la plus importante parmi les bactéries lactiques et leur activité estérasique est à l’origine de la production d’arômes fruités [44]. Lactobacillus helveticus et L. delbrueckii sont les espèces les plus utilisées dans les ferments lactiques et les cultures d’appoint.
1.2.2 La microflore bactérienne secondaire et la microflore d’affinage des fromages
Les bactéries ensemencées comme ferments d’affinage se retrouvent principalement à la surface des fromages [31]. Les bactéries corynéformes et psychrohalotolérantes sont les principaux groupes bactériens participant à l’affinage des fromages [45, 46]. Bien que leur présence soit confirmée depuis longtemps, leur rôle dans l’affinage n’est pas toujours bien connu.
Les bactéries corynéformes, dont l’observation microscopique révèle des bâtonnets de forme irrégulière, comprennent les genres Brevibacterium, Brachybacterium, Corynebacterium, Glutamicibacter et Microbacterium [31, 47, 48]. Bien que ces bactéries se retrouvent de façon omniprésente dans l’environnement, plusieurs espèces ont été isolées à partir de différentes variétés de fromages [31]. Les colonies de ce groupe de bactéries sont colorées (jaune, orange, beige ou rouge-beige). Les corynéformes se retrouvent, entre autres, à la surface des fromages Brick, Limburger, Romadour, Weinkase, Harzer Handkäse, Münster, Camembert, Appenzeller et Esrom [31, 49, 50].
Les bactéries du genre Brevibacterium sont aérobies, mésophiles, halotolérante (jusqu’à 15 % NaCl) et produisent des caroténoïdes (pigments jaunes et oranges) [51]. Brevibacterium linens est une bactérie dominante des fromages à croûte lavée, représentant jusqu’à 30 % de la microflore [31]. Ses enzymes protéolytiques et lipolytiques contribuent de façon déterminante à l’affinage des fromages Limburger, Münster, Brick, Tilsit et Appenzeller [31]. B. linens présente des activités peptidiques, estérolytiques et anti-Listeria [52]. Elle produit des composés volatils soufrés, comme le sulfure d’hydrogène, le méthanethiol et le sulfure de diméthyle en plus de l’ammoniaque [53, 54]. L’espèce
B. aurantiacum est impliquée dans l’affinage de plusieurs fromages irlandais, tels que le Gubbeen, l’Ardrahan et le Durrus [55].
Les espèces Brachybacterium tyrofermentans et B. alimentarium ont été isolées de la surface de fromages Gruyère et Beaufort [31]. Ces espèces sont anaérobies facultatives, halotolérantes (~15 % de NaCl) et leur croissance est optimale à 30 C à un pH de 7,3 [56]. Ces bactéries corynéformes, dont la croissance est optimale à une température entre 20 et 30 C, se distinguent par leur capacité à produire de la ménaquinone (vitamine K2) [56].
Les principales espèces de Corynebacterium répertoriées dans les écosystèmes fromagers sont C. variabile, C. casei, C. mooreparkense et C. flavescens. Les espèces appartenant à ce genre réalisent le catabolisme des acides aminés, possède des activités lipolytiques et protéolytiques et produit de l’ammoniaque ainsi que des composés sulfurés, tel que le méthanethiol [54]. Certaines espèces ont une activité estérase [31, 57]. Les Corynebacterium sp. produisent des colonies jaunes ou beige-jaune et sont aérobies facultatives [4, 50]. Dans l’environnement, on les retrouve dans le sol, à la surface des plantes et dans les eaux usées [31].
À ce jour, seules deux espèces de Microbacterium ont été retrouvées dans les fromages, soit Microbacterium lacticum, de couleur blanche-jaunâtre, et M. gubbeenense, de couleur beige ou beige-rouge [5, 49]. M. lacticum possède une activité anti-Listeria dans les fromages [58]. Ces espèces produisent de l’acide méthylbutyrique, de l’acide caproïque et du phényléthanol qui contribuent aux arômes des fromages [59]. Les Microbacterium sont généralement retrouvés en petite proportion et de façon sporadique à la surface des fromages à croûte lavée [49].
Les bactéries psychrohalotolérantes ont la propriété de croître en présence de sel (jusqu’à 8 %) et à des températures autour de 10 C [4, 46, 60]. Ce groupe comprend de nombreux genres qui sont généralement associés à des environnements marins, tels que Cobetia, Halomonas, Pseudoalteromonas et Psychrobacter [7, 8, 12, 45, 61]. La présence de ces bactéries à la surface des fromages peut s’expliquer par l’utilisation de solutions salines pour le salage (saumure) et les soins des fromages (solutions salines de lavage) [62]. Dans ce groupe, le genre le plus souvent impliqué dans l’affinage des fromages est Glutamicibacter (anciennement appelé Arthrobacter). Ce genre se retrouve dans le sol et
à la surface des fromages à croûte lavée. En présence de sel, cette bactérie aérobie et chimio-organotrophe est la première corynéforme à se développer puisqu’elle est halotolérante [31]. Les espèces du genre Glutamicibacter jouent un rôle majeur dans l’affinage des fromages Brick, Vacherin Mont d’Or et Tilsit [63, 64]. Certaines souches produisent des bactériocines, qui sont des peptides permettant d’inhiber la croissance de certaines bactéries indésirables [50, 54]. Les Glutamicibacter possèdent des activités lipolytiques et protéolytiques utiles au développement de la flaveur des fromages et produisent de l’ammoniaque. L’une des espèces les plus fréquentes, G. nicotianae, est de couleur jaune et représente environ 30 % des corynéformes présents dans les fromages [4]. D’autres taxons sont également connus, tel que G. arilaitensis, G. citreus, G. globiformis et G. variabilis, mais ceux-ci sont peu retrouvés dans les fromages [50]. Plusieurs genres appartenant au groupe des gamma-protéobactéries (Halomonas, Pseudoalteromonas et Psychrobacter) pourraient participer à l’apparition des propriétés sensorielles par leurs activités protéolytique et lipolytique. Ce groupe de bactéries est très commun et parfois très abondant dans plusieurs variétés de fromages français traditionnels [14]. Par exemple, le genre Psychrobacter est retrouvé chez l’Époisses [14]. Le genre Cobetia se retrouve dans les environnements laitiers [65] et contribue à l’écosystème des fromages Gorgonzola et de fromages à croûte lavée [61]. Le genre Halomonas est impliqué dans l’affinage des fromages Livarot [14]. Les bactéries psychrohalotolérantes se retrouvent aussi chez les fromages Caciotta [45].
1.2.3 La microflore fongique utile des fromages
Au-delà des bactéries, un grand nombre de champignons microscopiques (levures et moisissures) contribuent de façon importante au développement des fromages [32]. Les mycètes inoculés lors de la fabrication sont retrouvés à la fois dans la pâte et à la surface des fromages. Dans la pâte, ils sont présents à une concentration plus faible que les bactéries lactiques [29]. En surface, leur importance dépend de la variété de fromage produite [12]. Les principaux genres de mycètes retrouvés dans les fromages sont Geotrichum, Debaryomyces, Kluyveromyces et Penicillium.
La principale espèce de Geotrichum utilisée est G. candidum. Cette levure est dimorphique, c’est-à-dire qu’elle peut présenter un phénotype filamenteux (apparence blanche et duveteuse) ou levuriforme (colonies ayant l’apparence des « levures », beiges et crémeuses) [66]. G. candidum est souvent isolée d’environnements laitiers, bien qu’elle
ne métabolise pas le lactose [67]. Elle peut être utilisée comme ferment d’affinage autant dans les fromages à croûte fleurie qu’à croûte lavée. Elle est utilisée dans les fromages à pâte molle, comme le Camembert, le Limbourg, le Livarot et le Pont-l’Évêque, tout comme dans les fromages pressés comme le Saint-Nectaire [68]. Par sa capacité à cataboliser des acides aminés et des acides gras, elle possède un riche potentiel d’aromatisation par la production d’alcools primaires et secondaires, d’esters et leurs dérivés, d’acides gras volatils et de méthylcétones et de composés soufrés volatils en grandes quantités [4, 69]. G. candidum est très sensible au sel, un mauvais contrôle du salage pouvant entraîner un arrêt de sa croissance [70]. Les souches de G. candidum utilisent et produisent différents composés à différentes concentrations, ce qui en fait une levure dont le potentiel fromager est souche-dépendant [67].
Les levures du genre Debaryomyces peut assimiler le lactate, le citrate, le lactose et le galactose. Ces levures ont des activités lipolytiques et protéolytiques et ont la capacité de produire des composés aromatiques, tels que des méthylcétones, du 2-phényléthanol et du méthylthioacétate [71, 72]. Elles contribuent aussi à l’alcalinisation du caillé, ce qui favorise la colonisation de la surface des fromages par les bactéries désirables acidosensibles, tout en étant un compétiteur de microorganismes indésirables, tel que Clostridium sp. [71, 72]. Elles augmentent aussi la concentration d’aldéhyde à chaîne ramifiée. La principale espèce utilisée dans le domaine fromager est Debaryomyces hansenii. Cette levure a un rôle majeur dans l’affinage des fromages Danbo, Limburger, Romadour, Tilsit, Münster, Weinkäse, Harzer et de fromages bleus [73].
Kluyveromyces est un genre de levure qui produit une grande quantité de beta-galactosidase, une enzyme dont la fonction est de cliver le lactose, un disaccharide, en glucose et en galactose, des monosaccharides [74]. Elle est donc fréquemment utilisée dans la fabrication de fromage sans lactose [74]. Cette levure est aussi impliquée dans l’affinage du Cantal, du Cheddar, du Damietta, du Roquefort et du Camembert [74-76]. D. hansenii et K. marxianus sont souvent utilisées de pair, à cause de leur consommation complémentaire des métabolites; K. marxianus consomme d’abord le lactose, puis ensuite le lactate, alors que D. hansenii consomme les deux simultanément [32]. Ces mycètes entraînent l’hydrolyse des protéines et des lipides, ce qui permet l’implantation de la moisissure Penicillium dans les fromages à pâte molle après environ une semaine d’affinage [32].
Deux espèces du genre Penicillium sont particulièrement importantes en fabrication fromagère. P. camemberti est une moisissure d’apparence blanche et duveteuse qui donne l’aspect caractéristique des fromages à croûte fleurie. P. roqueforti est une moisissure de couleur bleue foncée qui est utilisée pour la fabrication de fromages à pâte persillée [4]. Ce genre de mycète produit une grande quantité de lipases extracellulaires, qui donne les caractéristiques des fromages à pâte molle tels que le Brie et le Camembert ou des fromages bleus tel que le Roquefort [77]. Ces moisissures métabolisent aussi les acides aminés et les acides gras, permettant la formation de nombreux arômes, dont les principaux sont des méthylcétones saturées, des alcools, des cétones, des acides gras courts et des composés soufrés [4, 78]. En deux à trois jours, P. camemberti contribue à l’alcalinisation de la surface des fromages, en transformant le lactate en CO2 [32].
1.2.4 La microflore dépendante du type de technologie fromagère
La microflore de la surface des fromages de spécialité est caractéristique du type de croûte (fleurie, lavée ou mixte) qui elle-même est le résultat du procédé de fabrication choisi. Chaque technologie fromagère implique des conditions de transformation, de moulage, de salage et d’affinage différentes, ce qui favorise également des communautés microbiennes diverses [12]. L’écosystème de surface correspond donc au type de fromage produit et influence l’apparence de ces derniers. Les fromages de spécialité les plus fréquents sont les fromages à croûte lavée, fleurie ou mixte [4] (Fig. 1).
Figure 1 Aspect visuel de fromages de spécialité. (A) Fromage à croûte lavée, de couleur orangée. (B) Fromage à croûte mixte, dont l’apparence est intermédiaire. (C) Fromage à croûte fleurie, dont la surface est blanche et duveteuse. Tiré de https://www.fromages.com/ [79].
1.2.4.1 Les fromages à croûte lavée
La couleur « orangée » caractéristique des fromages à croûte lavée (Fig. 1A) est le résultat de la croissance des bactéries corynéformes [4, 50]. Le procédé de fabrication de ces fromages implique l’application répétée de solution de lavage, saline ou non, et de brossage favorisant les bactéries psychrohalotolérantes [80]. La surface des fromages à croûte lavée est premièrement colonisée par des levures, en raison des conditions environnementales (faible pH, peu d’humidité relative, basse température des salles d’affinage et salinité
C
élevée). Celles-ci contribuent à l’alcalinisation de la surface, ce qui favorise l’implantation subséquente des bactéries corynéformes qui sont ensemencées lors de la fabrication et lors des soins des fromages ou encore qui proviennent de l’environnement. L’écosystème fongique en fin d’affinage a longtemps été considéré comme une contamination chez les fromages à croûte lavée, mais est maintenant mieux contrôlé [81]. Cette microflore est principalement composée des ferments d’affinage, plus particulièrement des genres Debaryomyces et Geotrichum [12, 14].
1.2.4.2 Les fromages à croûte fleurie
Les fromages à croûte fleurie (Fig. 1C) présentent généralement une moins grande diversité microbienne que les fromages à croûte lavée [12]. Dans les premiers jours de l’affinage, ces fromages présentent une couche de mycètes généralement composée des levures Kluyveromyces lactis, Saccharomyces cerevesiae, Debaryomyces hansenii et Geotrichum candidum et de la moisissure Penicillium camemberti [32]. Ces microorganismes contribuent à l’alcalinisation de la surface du fromage en consommant le lactate et permettent l’implantation de la microflore bactérienne. Les levures réalisent l’hydrolyse des protéines et des lipides, ce qui favorise la croissance de P. camemberti. Après une semaine d’affinage, cette moisissure recouvre entièrement la surface, ce qui donne l’apparence blanche et duveteuse caractéristique des fromages à croûte fleurie [70].
En raison de leur métabolisme aérobie et de leur croissance rapide dans les conditions utilisées pour l’affinage, G. candidum et les Penicillium, sont prédominantes à la surface des fromages à croûte fleurie [32]. À titre d’exemple, les conditions d’affinage pour le Camembert sont une température d’environ 12 C, une humidité relative de 90 % et un pH du caillé de 4,6-4,7 [32].
1.2.4.3 Les fromages à croûte mixte
La définition des fromages à croûtes mixte est difficile à établir puisque plusieurs interprétations sont retrouvées dans la littérature. Pour les fins de ce mémoire, la définition retenue est celle du concours des fromages du Québec, la Sélection Caseus. La définition des fromages à croûte mixte (Fig. 1B), aussi appelés fromages à croûte naturelle, correspond aux fromages « présentant en surface une flore bactérienne ou fongique diversifiée et dont l’entretien de la surface du fromage durant l’affinage peut varier » [82].
Habituellement, l’écosystème de la surface des fromages reflète la communauté microbienne présente dans l’environnement des fromages (et du lait cru, le cas échéant) et les microorganismes ne sont pas ensemencés avec un ferment d’affinage [83]. Ils présentent des caractéristiques intermédiaires se situant entre les fromages à croûte fleurie et à croûte lavée [12]. Leur surface présente généralement un enchevêtrement de mycélium blanchâtre, par la présence de Penicillium sp. et de Geotrichum sp., ainsi que des bactéries orangées appartenant aux genres Brevibacterium , Microbacterium et Glutamicibacter. [12].
1.2.5 La microflore et la production d’arômes
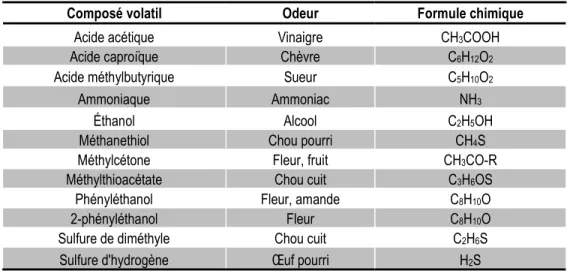
Les différents métabolites produits par la microflore sont à l’origine des caractéristiques organoleptiques des fromages. À ce jour, des centaines de composés volatils contribuant à l’arôme des fromages ont été isolés et identifiés [84]. Les principaux composés volatils impliqués dans l’affinage des fromages sont présentés au Tableau 1. Les fromages à croûte lavée sont généralement les plus odorants, principalement en raison des activités lipolytiques qui génèrent des acides gras à chaîne courte, mais aussi du fait de la dégradation des caséines par les espèces microbiennes en présence [85]. Les composés qui contribuent aux arômes de fromages sont largement étudiés dans la littérature scientifique [68, 75, 85-87].
Tableau 1 Odeur des composés volatils issus du métabolisme de la microflore fromagère.
Composé volatil Odeur Formule chimique Acide acétique Vinaigre CH3COOH Acide caproïque Chèvre C6H12O2 Acide méthylbutyrique Sueur C5H10O2 Ammoniaque Ammoniac NH3
Éthanol Alcool C2H5OH Méthanethiol Chou pourri CH4S Méthylcétone Fleur, fruit CH3CO-R Méthylthioacétate Chou cuit C3H6OS
Phényléthanol Fleur, amande C8H10O 2-phényléthanol Fleur C8H10O Sulfure de diméthyle Chou cuit C2H6S Sulfure d'hydrogène Œuf pourri H2S
1.3 La typicité fromagère et la notion de terroir
En 2003, le Groupe de travail sur les appellations réservées et les produits du terroir, appelé Rapport Desjardins, a donné la définition suivante en ce qui concerne les produits du terroir :
Un produit du terroir est un produit qui provient - ou dont les principales composantes proviennent - d'un territoire délimité et homogène et dont les caractéristiques qui le distinguent de façon significative des produits de même nature reposent sur la spécificité de ce territoire. Ses caractéristiques dépendent à la fois des particularités du milieu, comme la géologie, le climat, le relief, la culture, l'histoire ainsi que du savoir et du savoir-faire, traditionnels ou émergents, et de ses habitants [88].
Outre les procédés de fabrication, l’environnement de la ferme laitière semble avoir un impact sur le développement des fromages [89]. La composition du lait varie en fonction de la race d’animaux, de la gestion du troupeau ou du niveau d’hygiène de l’environnement de production [90, 91]. Le moyen de transport du lait jusqu’au lieu de transformation, le moment de production dans la journée, la saisonnalité et l’emplacement géographique influencent aussi la composition microbiologique du lait [25, 92, 93]. L’environnement, autant à la ferme qu’à la fromagerie, module la microflore des fromages. Tous ces paramètres contribuent à la typicité des fromages, c’est-à-dire l’ensemble des caractéristiques qui en font ses particularités. Lorsqu’un produit est typique d’un savoir-faire ou d’une région, il peut alors porter une appellation contrôlée, garantissant sa réputation. Au Québec, on compte six appellations réservées et reconnues, dont une mettant en valeur les fromages de vache de race Canadienne [94].
L’évaluation de la typicité passe par la caractérisation de la microflore. En 2011, une étude d’envergure a été menée sur la caractérisation fongique et chimique des laits servant à la fabrication de fromages du terroir québécois [95]. Les analyses ont permis de recenser 82 espèces de levures et de moisissures à partir de plus d’une centaine d’échantillons de lait cru de vache. Une persistance des microorganismes a été observée au cours des sept mois de prélèvement [16]. Différentes méthodes peuvent être utilisées afin d’identifier la communauté microbienne, chacune ayant des avantages et des limitations [10, 96, 97].
1.4 Les méthodes d’identification du microbiote fromager
1.4.1 Les analyses phénotypiques de microorganismes isolés
L’analyse phénotypique se fait principalement en microbiologie classique, par la mise en culture des microorganismes se retrouvant dans le produit à caractériser. Bien que les bactéries et les mycètes les plus abondants au sein des écosystèmes fromagers peuvent
être recensés par cette technique, une grande proportion de microorganismes viables ne sont pas cultivable en laboratoire, ce qui peut constituer un frein à cette approche [10, 98]. On distingue deux types d’analyses phénotypiques. Le premier regroupe les analyses morphologiques qui s’intéressent à l’apparence (couleur, taille, forme) d’une culture isolée sur milieu de culture solide [10]. Le deuxième, les tests biochimiques, servent à tester les propriétés métaboliques d’un microorganisme en culture et d’observer sa capacité à utiliser et transformer un milieu sélectif (ex. gélose sang, gélose MacConkey, tests enzymatiques, tests de fermentation) à des fins d’identification [96]. Ces tests sont généralement associés à une clé dichotomique et les résultats obtenus permettent de progresser dans la clé pour obtenir une identification [99].
1.4.2 Les analyses moléculaires non ciblées
Les analyses par biologie moléculaire s’intéressent à l’étude de l’ADN (génomique) ou de l’ARN (transcriptomique). La métagénomique permet d’identifier la composition d’un écosystème et de connaître l’abondance (absolue ou relative) des microorganismes présents [100]. Pour sa part, la métatranscriptomique permet de connaître l’expression des voies métaboliques sollicitées dans des conditions spécifiques, et de connaître le niveau d’activité métabolique des microorganismes présents dans un écosystème [101]. Ces méthodes ont l’avantage de demander moins de temps et d’énergie que les méthodes nécessitant l’isolement, la culture et la caractérisation d’isolats microbiens [102]. Les méthodes non ciblées d’identification moléculaire d’ADN se divisent en deux groupes : le typage moléculaire et le séquençage d’ADN.
Le typage moléculaire, aussi appelé empreinte génétique (en anglais, DNA fingerprinting), a été mis au point en 1985 par Alec Jeffreys [103]. Cette méthode implique la digestion enzymatique de l’ADN pour ensuite en comparer le polymorphisme (comparaison de la taille des fragments d’ADN, du nombre et de la distance entre les sites de restriction). L’analyse du polymorphisme de longueur des fragments de restriction terminaux (T-RFLP – en anglais, terminal restriction fragment-length polymorphism) est une approche rapide et sensible utilisée pour distinguer les souches d’une même espèce [97]. Le T-RFLP peut être utilisé pour caractériser des écosystèmes diversifiés, comme celui des fromages [102, 104]. La principale limitation de cette méthode est qu’elle ne permet pas d’accéder facilement à l’identification taxinomique des microorganismes détectés. Également, la résolution obtenue pour un écosystème donné dépend de la présence de sites de restriction correspondant à
l’endonucléase utilisée. Il est donc possible que certains microorganismes ne soient pas discriminés par manque de sites de restriction dans la région d’intérêt de leur génome [102].
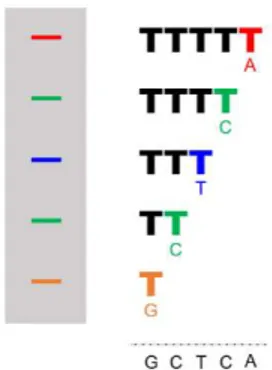
La technologie du séquençage de l’ADN a vu le jour dans les années 70 avec la méthode de Sanger et est en constante évolution depuis [105]. Cette réaction utilise une ADN polymérase et une amorce d’oligonucléotides afin de cibler une région spécifique à séquencer. L’ADN double brins ciblé pour le séquençage est d’abord séparé en ADN simple brin par chauffage puis polymérisé à partir d'une amorce, par l’ajout de désoxyribonucléotides (dNTP). La réaction de séquençage implique aussi une polymérisation en présence d’un mélange de dNTP et de didésoxyribonucléotides (ddNTP) fluorescents dont la longueur d’onde d’émission diffère selon le nucléotide (ddATP, ddGTP, ddCTP ou ddTTP). L’incorporation de ddNTP entraîne un arrêt prématuré et aléatoire de la polymérisation, ce qui génère des fragments de toutes les tailles qui contiennent un nucléotide fluorescent à l’extrémité 3’ (Fig. 2). Ensuite, la séparation des fragments selon leur taille est effectuée par électrophorèse capillaire, et la détection de la fluorescence émise permet de déterminer la séquence du brin d’ADN d’intérêt par l’identification successive des ddNTP détectés (Fig. 2).
Figure 2 Schématisation du séquençage de Sanger. (À gauche) Représentation de la migration des fragments générés à la suite de la réaction de polymérisation sur gel d’électrophorèse capillaire et détection de la couleur associée à chaque ddNTP. (À droite) Représentation du fragment synthétisé par polymérisation à la suite de l’arrêt lors de l’incorporation d’un ddNTP à l’extrémité 3’ et déduction de la séquence d’ADN résultante. Figure adaptée de Valencia [106].
Le séquençage permet d’analyser quelques centaines de paires de bases (pb), alors que les génomes en font plusieurs millions. À titre d’exemple, la taille du génome de la levure Geotrichum candidum est d’environ 25 Mb [107]. Les premières approches de séquençage consistaient à fractionner le génome et à générer des banques de clones contenant chacun un fragment différent du génome de l’organisme à séquencer. Pour y arriver, l’ADN est
extrait, fragmenté par une endonucléase de restriction, puis intégré dans un vecteur de clonage (chromosome artificiel bactérien, de levure, humain ou dérivé du phage). Les constructions sont mises en culture, avant de sélectionner par hybridation les clones recombinants. Ensuite, chaque clone peut être séquencé, puis les séquences sont assemblées, pour obtenir le génome complet. Les technologies de séquençage ont connu depuis une véritable révolution. Ainsi, le coût du séquençage a chuté considérablement alors que les performances des appareils et des méthodes se sont perfectionnées, augmentant ainsi son accessibilité et ses performances [108]. À titre d’exemple, le séquençage d’un génome humain (3,2 Gb) est passé de 100 M$ au début des années 2000 à moins de 5000 $ aujourd’hui avec les techniques de séquençage de nouvelle génération [109, 110].
Au tournant du XXIe siècle est apparu le séquençage de nouvelle génération (NGS – en anglais, next generation sequencing). Comme cette technologie génère une grande quantité de données, le terme de séquençage à haut débit (HTS – en anglais, high-throughput sequencing) est souvent utilisé. Différentes compagnies ont développé des technologies dont les applications varient selon la méthodologie employée (Tableau 2). Il est possible de reconstituer le génome de microorganismes complexes, d’analyser des produits de PCR (amplicons), de détecter des mutations et plus encore [111].
La fabrication de banque de clones a été remplacée par la construction de librairies sur supports solides directement à partir de l’ADN isolé d’un microorganisme ou d’une communauté microbienne en utilisant des adaptateurs, ce qui implique que la mise en culture d’une multitude de clones n’est plus nécessaire, demandant donc beaucoup moins de temps, de ressources et de travail [112]. Les réactions de séquençage ont été parallélisées, afin d’analyser plusieurs échantillons à la fois. Finalement, le système d’acquisition du signal lors du séquençage se déroule pendant la réaction elle-même évitant le recours à une technique d’électrophorèse capillaire, ce qui permet une automatisation de l’acquisition des données [111].
Tableau 2 Comparaison des technologies de NGS. Tableau adapté de Mardis [113].
Non pairés = En anglais, single end; Pairés = En anglais, paired end.
1.4.3 Le séquençage du génome complet d’un organisme
Afin de réaliser le séquençage de l’ensemble du génome d’un organisme, l’ADN extrait est fragmenté, ligaturé à des adaptateurs, fixé à la plaque de séquençage, dénaturé, puis séquencé. Lors du cycle de séquençage, les nucléotides fluorescents sont incorporés un à un. À chaque incorporation de nucléotide, la fluorescence émise est captée et interprétée par une caméra, ce qui permet de déduire la séquence des brins d’ADN (Fig. 3). La caractérisation de l’ensemble du génome d’un microorganisme permet des identifications précises. Cependant, le génome présentant plusieurs millions de paires de bases, il s’agit d’un procédé coûteux. La technologie Illumina est l’une des plus utilisées pour le séquençage de génomes complets. Une alternative intéressante pour l’identification des microorganismes est l’utilisation du barcoding.
Compagnie Longueur du séquençage Applications
454/Roche 400 pb (non pairés) Génomes bactériens et viraux, séquençage massif d’amplicons, validation de mutations ponctuelles, détection ciblée de mutations somatiques.
Illumina 150–300 pb (pairés)
Génomes complexes (humains, souris et plantes) et applications de NGS à l'échelle du génome, séquençage de l'ARN, produits de capture par hybridation ou séquençage massif d’amplicons, détection de mutations somatiques, expertise médico-légale, tests prénataux non invasifs.
ABI SOLiD 75 pb (non pairés, ou 50 pb pairés)
Applications génomiques complexes (humaines, souris, plantes) et NGS à l'échelle du génome, séquençage de l'ARN, produits de capture par hybridation ou séquençage massif d’amplicons, détection de mutations somatiques. Pacific Biosciences Jusqu'à 40 kb (non pairés ou séquence circulaire consensus)
Génomes complexes (humains, souris et plantes), microbiologie et génomes de maladies infectieuses, détection de transcrits de fusion, détection de méthylation.
Ion Torrent 200–400 pb (non pairés) Séquençage massif d’amplicon, microbiologie et maladies infectieuses, détection de mutations somatiques, validation de mutations ponctuelles.
Oxford Nanopore Variable: dépend de la préparation de la librairie (1D – non pairés, ou 2D - pairés)
Surveillance des agents pathogènes, détection de mutations ciblées, métagénomique, génomes bactériens et viraux.
Qiagen
Figure 3 Technologie de séquençage à haut débit d’Illumina. (À gauche) Simple brin d’ADN fixé à la plaque par un adaptateur, auquel une amorce est hybridée afin de débuter l’élongation, en présence de nucléotides terminateurs réversibles fluorescents. (Au centre) À la suite de l’ajout du nucléotide, un signal lumineux est émis et la fluorescence est interprétée par une caméra. (À droite) Cette opération se réalise en parallèle à plusieurs endroits sur la plaque. L’ensemble des signaux est compilé afin de connaître la séquence de chaque fragment. Figure adaptée de Chaitankar et al. [114].
1.4.4 Le barcoding
Le barcoding, aussi appelé séquençage d’amplicons, est une méthode qui a fait son apparition au début des années 1990 [115]. Il s’agit d’une méthode de biologie moléculaire fiable, rapide et peu coûteuse qui est utilisée pour identifier un microorganisme isolé [116, 117]. Elle consiste à cibler une courte séquence d’ADN, de quelques centaines de paires de bases (représentative du genre, de l’espèce ou de la souche, le plus souvent l’ADNr), afin de la comparer à une banque de données, ce qui mène à son identification [117].
1.4.5 Le métabarcoding
Le métabarcoding, ou séquençage massif d’amplicons, est une utilisation à grande échelle du barcoding qui est appliqué dans un écosystème complexe. Il consiste à extraire l’ADN total des microorganismes présents dans un mélange et à séquencer en grand nombre les marqueurs universels permettant de les identifier (le plus souvent l’ADN ribosomal, voir 1.4.5.1). Cette méthode est utilisée afin d’identifier des milliers de microorganismes à la fois et est régulièrement utilisée pour étudier des écosystèmes complexes, tels que les fromages [12, 14, 25, 118-121]. L’émergence de cette approche d’analyse des écosystèmes microbiens a été rendue possible grâce au développement du NGS [122]. Ces études ont permis d’identifier la microflore bactérienne et/ou fongique de plusieurs fromages à travers le monde. Cette caractérisation permet de mieux comprendre le fonctionnement de
communauté microbienne complexe [12], d’étudier l’évolution des microorganismes au cours de l’affinage et le rôle qu’il joue dans la fabrication fromagère [6, 14, 93, 119-121, 123, 124].
1.4.5.1 Le choix de la région cible de métabarcoding
La région utilisée afin d’identifier les microorganismes doit respecter plusieurs critères. Premièrement, elle doit être ubiquitaire, c’est-à-dire présente chez tous les individus de la communauté [125]. Des gènes impliqués dans des activités métaboliques essentielles sont généralement utilisés [126]. Deuxièmement, la région cible doit comporter une ou des portions variables, à des fins de discrimination, et des portions conservées, afin de pouvoir cibler la région avec des amorces universelles [127]. Plusieurs régions correspondent à ces deux critères, mais les plus fréquemment utilisées font partie de l’ADNr 16S chez les bactéries et des espaceurs de transcription interne de l’opéron des ADNr, les ITS (en anglais, Internal Transcribed Spacer) chez les mycètes [128]. Troisièmement, la caractérisation d’écosystèmes par métabarcoding nécessite le séquençage d’une courte séquence, qui doit pouvoir permettre l’identification des microorganismes à un niveau taxinomique raisonnable, par exemple le genre. Puisque des milliers de microorganismes seront séquencés, il devient important de cibler une courte région [129]. L’utilisation de la technologie MiSeq d’Illumina permet une longueur maximale de 300 pb et est généralement utilisé en mode pairé pour couvrir jusqu’à 500 pb, ce qui est compatible aux besoins du métabarcoding [130].
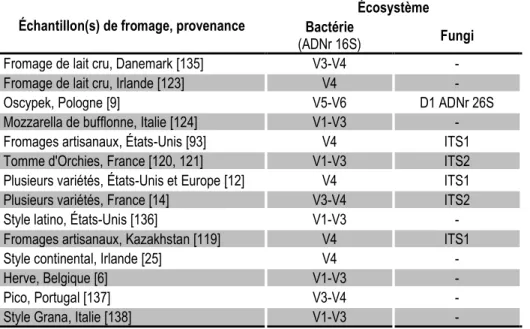
La sélection d’un locus à utiliser en métabarcoding qui demeure représentatif de l’écosystème complet demeure difficile à choisir à ce jour [117]. La meilleure région à séquencer au sein de l’ADNr 16S et les ITS est d’ailleurs un sujet de débat dans la littérature scientifique [117, 131-134]. Au cours des dernières années, plusieurs études de métabarcoding étudiant des écosystèmes fromagers ont été réalisées à l’aide de différentes régions cibles (Tableau 3).
Tableau 3 Régions cibles de l’ADNr utilisées pour étudier les écosystèmes fromagers.
Échantillon(s) de fromage, provenance
Écosystème Bactérie
(ADNr 16S) Fungi Fromage de lait cru, Danemark [135] V3-V4 - Fromage de lait cru, Irlande [123] V4 - Oscypek, Pologne [9] V5-V6 D1 ADNr 26S Mozzarella de bufflonne, Italie [124] V1-V3 - Fromages artisanaux, États-Unis [93] V4 ITS1 Tomme d'Orchies, France [120, 121] V1-V3 ITS2 Plusieurs variétés, États-Unis et Europe [12] V4 ITS1 Plusieurs variétés, France [14] V3-V4 ITS2 Style latino, États-Unis [136] V1-V3 - Fromages artisanaux, Kazakhstan [119] V4 ITS1 Style continental, Irlande [25] V4 - Herve, Belgique [6] V1-V3 - Pico, Portugal [137] V3-V4 - Style Grana, Italie [138] V1-V3 -
Chez les bactéries, l’ADNr 16S est d’une taille approximative de 1500 paires de bases (pb) et comprend neuf régions hypervariables (Fig. 4A) [139]. Jusqu’ici les régions hypervariables V3-V4 (460 pb) et V6-V8 (300 pb) ont été utilisées avec succès pour décrire des écosystèmes fromagers et laitiers [14, 135, 137, 140, 141]. L’équivalence entre celles-ci n’a cependant pas été établie à ce jour.
Chez les mycètes, les régions ITS1 et ITS2 sont séparées d’environ 80 pb par le gène de l’ADNr 5.8S (Fig. 4B). Les régions ITS sont des régions non-codantes d’une longueur variable, soit d’environ 600 ± 100 pb [128]. L’ITS1 compte environ 200 pb et est flanqué par l’ADNr 18S en amont et l’ADNr 5.8S en aval. L’ITS2 compte environ 300 pb et est situé en aval de l’ADNr 5.8S et en amont de l’ADNr 26S [142, 143]. Les deux régions (ITS1 et ITS2) sont régulièrement utilisées pour caractériser les écosystèmes fongiques fromagers [12, 14, 93, 119, 120]. Ici également, l’équivalence entre ces deux régions n’a pas été étudiée.
Figure 4 Représentation des cibles de l’ADNr bactérien (A) et fongique (B) utilisées en métabarcoding. (A) L’ADNr 16S bactérien comporte neuf régions hypervariables, numéroté de V1 à V9. (B) L’ADNr fongique présente deux espaceurs de transcription interne, ITS1 et ITS2. Figure adaptée de Chakravorty et al., 2007 [139] et Porras-Alfaro et al., 2013 [143].
L’absence de consensus sur la région cible à utiliser lors de l’analyse par métabarcoding rend difficile la comparaison entre les études. Le choix de la région a un rôle important dans la description des écosystèmes puisque la diversité génétique varie d’un locus à un autre [133], ce qui influence la performance d’identification. Cela implique que pour différentes régions, le recensement des espèces ne sera pas identique. Selon l’environnement étudié et la composition de la communauté microbienne, la meilleure région cible peut varier.
1.5 La notion d’unité taxinomique opérationnelle
L’unité taxinomique opérationnelle (UTO; en anglais OTU pour operational taxonomic unit) est une unité introduite en 1963 par Robert R. Sokal et Peter H. A. Sneath qui désigne le regroupement d’organismes sur la base de leur similarité pour une région d’ADN donnée [144]. Les séquences sont regroupées selon leur seuil de similarité entre elles, choisi par l’utilisateur. Depuis 1994, le seuil de 97 % d’identité nucléotidique est utilisé pour regrouper des séquences appartenant à la même espèce [145]. Ainsi, un seuil de 97 % est généralement utilisé lors des études d’écologie microbienne, c’est-à-dire que les séquences présentant un seuil de similarité supérieur ou égal à 97 % sont regroupées et présenteront la même espèce [146]. Ce seuil correspond approximativement au seuil de similarité d’une espèce pour l’ADNr 16S et est similaire pour les ITS [147]. La notion d’UTO se distingue de la définition d’espèce, puisqu’elle ne réfère aucunement aux caractéristiques morphologiques des individus. Elle est uniquement basée sur la similarité des séquences utilisées. V1 V2 V3 V4 V5 V6 V7 V8 V9 ITS1 ITS2 18S 5.8S 26S A B
![Tableau 2 Comparaison des technologies de NGS. Tableau adapté de Mardis [113].](https://thumb-eu.123doks.com/thumbv2/123doknet/3142679.89475/31.918.126.781.136.578/tableau-comparaison-technologies-ngs-tableau-adapté-mardis.webp)

![Tableau 4 Caractéristiques des outils pour le nettoyage des séquences Illumina. Traduit et adapté de Del Fabbro et al [163]](https://thumb-eu.123doks.com/thumbv2/123doknet/3142679.89475/39.918.130.767.232.751/tableau-caractéristiques-nettoyage-séquences-illumina-traduit-adapté-fabbro.webp)