Effect of stress on vacancy formation and diffusion in fcc systems: Comparison between DFT calculations and elasticity theory
15
0
0
Texte intégral
Figure
![Fig. 1. From left to right: detail of atomic jumps for hydrostatic ( p ), [001] ( σ zz ) and [111] ( σ 111 ) uni-axial stresses, and for shear stress in plane [010] ( σ xy )](https://thumb-eu.123doks.com/thumbv2/123doknet/2942373.79308/6.892.80.840.85.293/right-atomic-jumps-hydrostatic-axial-stresses-stress-plane.webp)
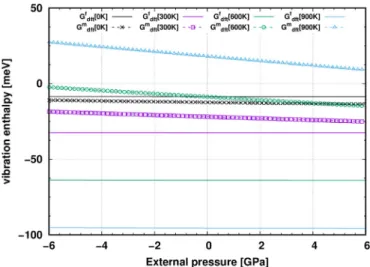
+5


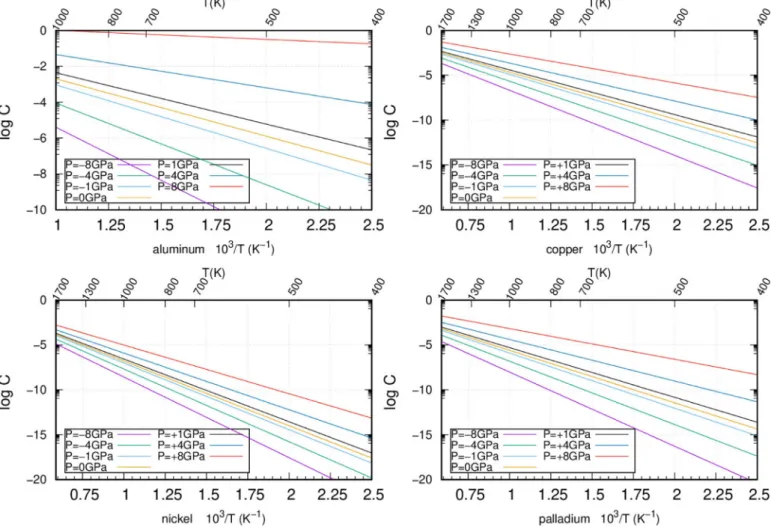
![Fig. 9. Evolution of the formation (top) and migration (middle) enthalpies as a function of pressure, under a uni-axial stress along [111], σ 111 (in GPa)](https://thumb-eu.123doks.com/thumbv2/123doknet/2942373.79308/12.892.69.438.81.852/evolution-formation-migration-middle-enthalpies-function-pressure-stress.webp)

Documents relatifs