Biogeochemical processes and buffering capacity concurrently affect acidification in a seasonally hypoxic coastal marine basin
Texte intégral
Figure
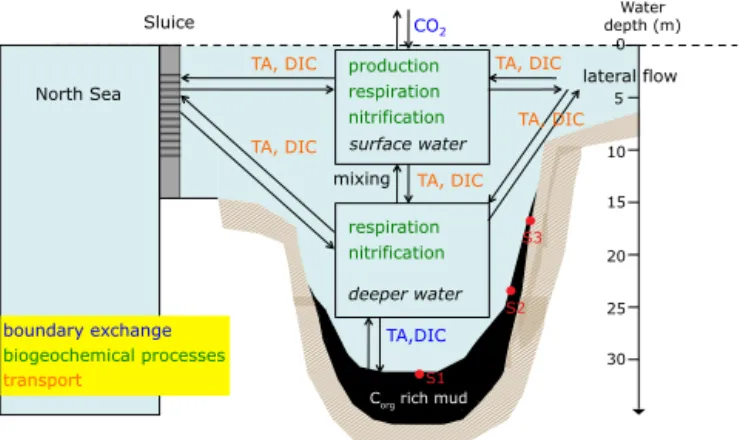

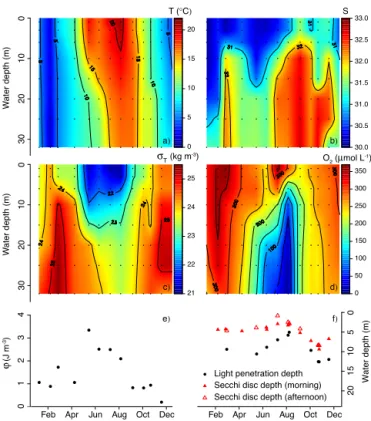
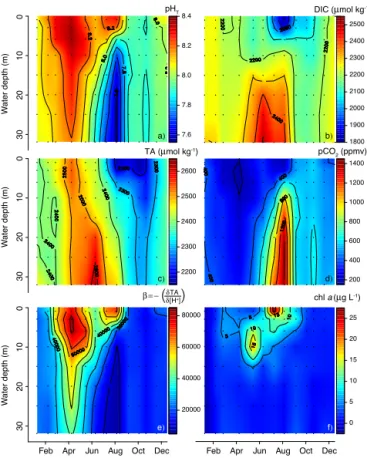
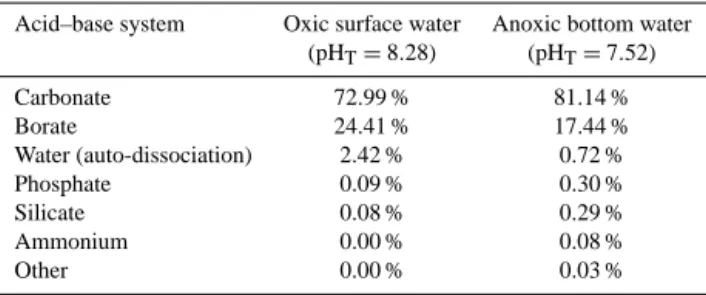
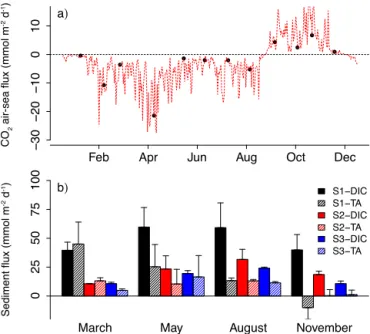
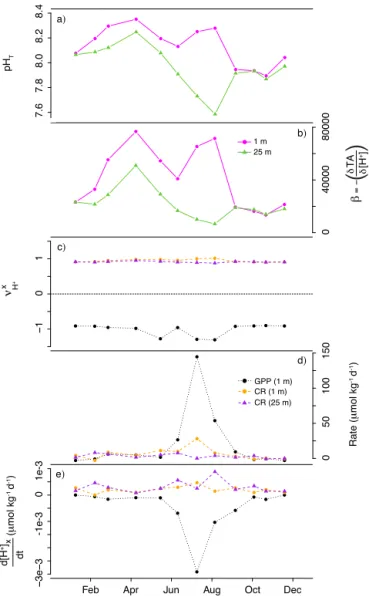

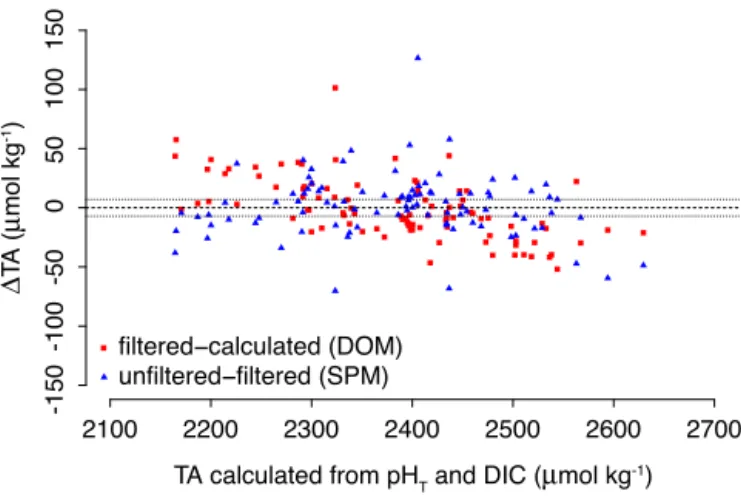
Documents relatifs
Masinga Dam (a large reservoir on the upper river) showed a strong retention of nutrients and an uncoupling of DOC and POC: while DOC pools and d13C signatures were similar above,
Thus, impaired recognition of facial expressions of disgust reported in some studies with adults (Sprengelmeyer et al., 1997a; Corcoran et al., in press) do not appear to extend
Power Control received frames Power Control sent frames No Power Control received frames No Power Control sent frames Theoretical model (empirical distribution) Theoretical
L’intruse, c’est aussi Monsieur Krebs, c’est-à-dire le cancer. Entre récit romanesque et réflexion philosophique, Gilbert Pingeon aborde ce thème avec la lucidité et
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
Knowing that Claytec panel, Claytec slimcoat and tested clay-based plasters are realised with the same clay, a tendency for the evolution of thermal conductivity
L’équilibre entre lecture historique, analyse textuelle et réflexion théorique est en général exemplaire dans chacun des textes, qui n’hésitent pas à quitter le domaine clos
• Samedi 24 et dimanche 25 : portes ouvertes : programme spécial proposé au public (aussi large que possible), deux journées intenses de cinéma, pas moins de
