REDD1 deficiency protects against nonalcoholic hepatic steatosis induced by high‐fat diet
Texte intégral
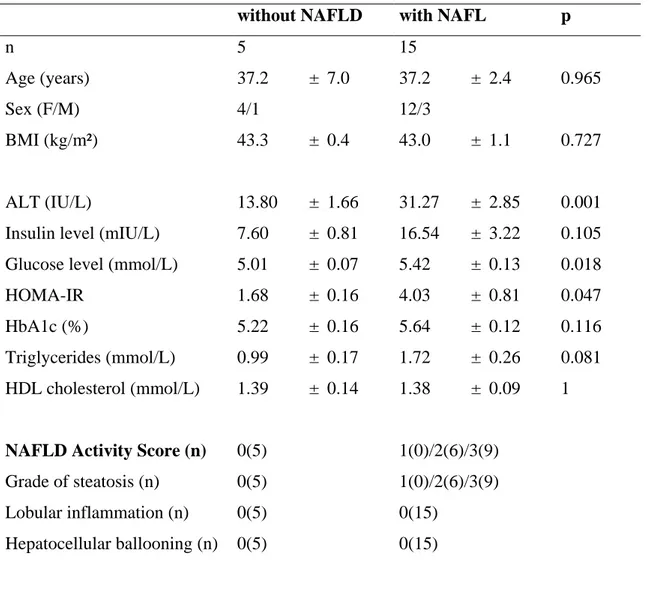
Figure
Documents relatifs
PARP2 deficiency affects invariant-NKT,-cell maturation and protects mice from ,Concanavalin A- induced liver injury.... 1 PARP2 deficiency affects invariant-NKT-cell maturation
We assessed the probable impact of chlordecone on the progression of liver fibrosis in the population of contaminated areas, by developing a mouse model of chronic co-exposure
Moderate chronic ethanol consumption exerts beneficial effects on nonalcoholic fatty liver in mice fed a high-fat diet possible role of higher formation of triglycerides enriched
Le Conseiller d'Etat Ducotterd, directeur militaire, remercie d'une solide poignée de mains ce soldat, et à travers lui tant d'autres, leur exprimant la reconnaissance de
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE
The semantics of temporal formulae in JTPL and translation rules into JML annotations are detailed in [28] for safety properties and in [1] for liveness properties.. This section
We present a girl with asymptomatic liver herniation through a postoperative right hemidiaphragm defect related to the implantation of an ascending aorta to upper abdominal
Indeed, we demonstrate that taurine treatment in mice fed with a HFD decreased the overall levels of plasma insulin during 24 h; it also decreased the insulin levels during the
