Conception et réactivité de complexes mono- et polymétalliques d'éléments f en bas degré d'oxydation
Texte intégral
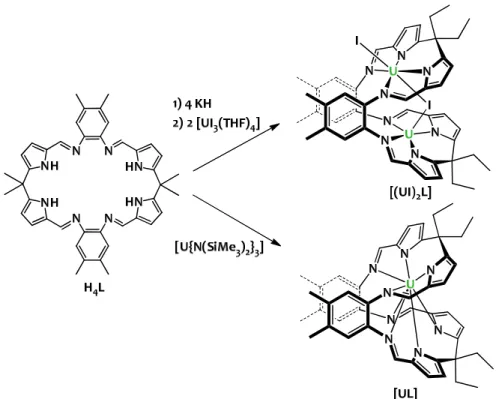
Figure
![Figure I.9. Mercury diagram of the solid-state molecular structure of the bridging carbonate uranium complex [{(( t-Bu ArO) 3 mes)U} 2 (μ-η 2 :η 2 -CO 3 )]](https://thumb-eu.123doks.com/thumbv2/123doknet/12705513.355826/41.892.178.735.111.473/figure-mercury-diagram-molecular-structure-bridging-carbonate-uranium.webp)
![Figure I.12. Mercury diagrams of the solid-state molecular structures of the uranium nitrido clusters ([U 4 (μ 4 -N)(μ-1,1-N 3 ) 8 (CH 3 CN) 8 I 6 ][(Cs(CH 3 CN) 3 ]) n (left) and [U(Cp*)(μ-I) 2 ] 3 (μ 3 -N) (right)](https://thumb-eu.123doks.com/thumbv2/123doknet/12705513.355826/48.892.204.711.475.759/figure-mercury-diagrams-molecular-structures-uranium-nitrido-clusters.webp)
![Figure I.15. Mercury diagram of the solid-state molecular structure of the U(V) terminal nitride [U(N)(Tren TIPS )] -](https://thumb-eu.123doks.com/thumbv2/123doknet/12705513.355826/52.892.122.791.326.665/figure-mercury-diagram-solid-molecular-structure-terminal-nitride.webp)
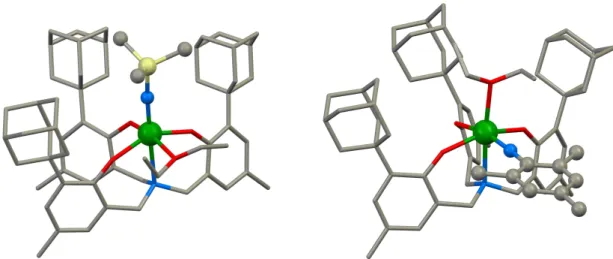
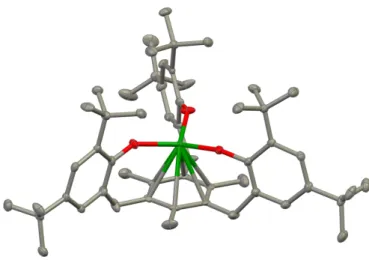
![Figure II.9. Solid-state molecular structure of the two isomeric forms of [U 2 (µ-salophean){N(SiMe 3 ) 2 } 5 ] 5-a (top) and 5-b (bottom)](https://thumb-eu.123doks.com/thumbv2/123doknet/12705513.355826/85.892.234.681.109.690/figure-solid-state-molecular-structure-isomeric-forms-salophean.webp)
![Figure II.17. Solid-state molecular structure of [Na(18c6)(py) 2 ] 2 [U(bis-salophen) 2 ] 7-18c6](https://thumb-eu.123doks.com/thumbv2/123doknet/12705513.355826/94.892.246.674.118.439/figure-solid-state-molecular-structure-na-bis-salophen.webp)
Documents relatifs
In this paper we have introduced the concept of "Social Radio", a platform for information broadcasting in online communities. Online communities' members
Figure 6 shows volumetric phase assemblages calcu- lated using GEMS for binder systems containing OPC + 15% CAC1 or OPC + 15% CAC2 (by mass) in lime- stone deficient or limestone
The method we have selected to characterize the prompt multiplication properties of the sub-critical assembly is based on the measurement of the decay of the neutron population
In the conventional picture of reactants di ffusing in the ice mantle and meeting each other to form iCOMs, the bulk chemistry at low or mild (before the ice desorption)
Due to the less important reputation of Vin- cenzo Capello compared with the Andrea Doria’s fame (the still powerful captain when Gio- vio writes his praises), Giovio takes advantage
Over the last decade, the ferrous derivative of Alcaligenes xylosoxidans cytochrome c´ (Axcyt c’) and of cardiolipin-bound horse heart cytochrome c (CL-hhcyt c) have
and Intensive Care, CHI, CRC, Créteil, France (Durrmeyer); Department of Neonatal Pediatrics, Sud Francilien Hospital, Evry, France (Granier); Neonatal intensive care unit, Robert
The challenges to the future include: continuing to develop strategies for promoting HCI education in computer science programs; make national conferences attractive for
