HAL Id: tel-00011846
https://tel.archives-ouvertes.fr/tel-00011846
Submitted on 8 Mar 2006
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
de sélénium
Gérard Gouedard
To cite this version:
Gérard Gouedard. Etude par pompage optique de l’état B3 de la molécule de sélénium. Physique
Atomique [physics.atom-ph]. Université Pierre et Marie Curie - Paris VI, 1984. Français.
�tel-00011846�
LABORATOIRE
DE
PHYSIQUE
DE
L’ÉCOLE
NORMALE
SUPÉRIEURE
THESE
DEDOCTORAT
D’ETAT
ès Sciences
Physiques
présentée
à
L’Université Pierre
et Marie CURIE
(Paris
VI )
par
Gérard
GOUEDARD
pour
obtenir
le
grade
de
Docteur
ès Sciences
Sujet
de la
Thèse : "ETUDE
PAR POMPAGE
OPTIQUE
DE
L’ETAT
B
3
03A3
-
u
DE
LA MOLECULE DE
SELENIUM".
Soutenue
le
Janvier
1984
Devant
le
Jury
composé
de :
M.
J. BROSSEL
)
Président
Mme H. LEFEBVRE-BRIONM.
J.C.
LEHMANNExaminateurs
M.J.C. PEBAY-PEYROULA
M.
J. VIGUE
M.
R.F.BARROW
)
.Invité
présentée
àL’Université Pierre et Marie CURIE
( Paris VI )
par
Gérard GOUEDARD
pour obtenir le
grade
de Docteur ès SciencesSujet
de la Thèse : "ETUDE PAR POMPAGEOPTIQUE
DE L’ETAT B3
03A3
-
u
DE LA MOLECULE DE SELENIUM".
Soutenue le Janvier 1984
Devant le
Jury
composé
de :M. J. BROSSEL
)
Président Mme H. LEFEBVRE-BRION M. J.C. LEHMANN Examinateurs M. J.C.PEBAY-PEYROULA
M. J. VIGUE)
M. R.F. BARROW)
Invitéenvironnement
scientifique
et
humain
exceptionnel.
Jean-Claude
LEHMANN
aguidé
mespremiers
pas dans
la
recherche
et,
malgré
sestrès
nombreuses
occupations,
atoujours
suivi
montravail
avecla
plus
grande
attention. Je lui
exprime
ici
maprofonde gratitude.
Madame
H.
LEFEBVRE-BRION
et
Monsieur
J.C.
PEBAY-PEYROULA
ont
accepté
de
faire
partie
de
cejury
de
thèse ;
je
les
enremercie
et
suis très
honoré
de
cette
marque d’intérêt.
Au
coursde
cetravail,
j’ai
eude nombreux
contacts
avecle
Profes-seur
BARROW,
avecqui j’ai
pu
collaborer dans d’excellentes conditions.
Je
lui
ensuis très
reconnaissant,
ainsi que
d’avoir
accepté
de
s’expatrier
momentanément
pour
participer
à
ce jury.
Je remercie très vivement A. JENOUVRIER
pour
m’avoir
communiqué
sesrésultats
avant
publication,
et
pour les
fructueuses
discussions
que
nous avons eues.J’ai
profondément
apprécié
l’ambiance de
notre
groupe
de
recherche ;
nous avans
toujours
pu
composer
uneéquipe
unie
et
solidaire,
tant
auniveau
expérimental
que
théorique.
Le
Professeur
F.W.
DALBY
aréalisé
auLaboratoire
les
premières
études
d’effet
Hanle
surla
molécule
de sélénium,
et
m’a
permis
de
les
poursuivre ; je
lui
exprime ici
toute
magratitude.
Je remercie
vive-ment
Michel
BROYER pour les
nombreuses
et
fructueuses
discussions
que
nous avons euesdans
la
phase
initiale
de
cetravail. Nicolas BILLY n’a
ménagé
ni
sontemps,
ni
sapeine
pour m’aider
lors
de
la rédaction
de
cemémoire,
je
lui
ensuis
profondément
reconnaissant. J’ai
bénéficié
de
façon plus
continue
encore
du soutien de
Jacques
VIGUE.
Je
voudrais
qu’il
sache combien
sesgrandes
capacités scientifiques,
sonenthousiasme
et
sonamitié
m’ont
été
précieux.
Il
ade
plus accepté
de
faire partie
de
cejury, je
l’en remercie
très
vivement.
Les
expériences
décrites
ici
ont
été réalisées dans la
partie
Halle
aux
Vins
du Laboratoire. Je voudrais que
tous
mescamarades sachent
combien
j’ai apprécié d’ y
travailler
enleur
compagnie ;
tous
m’ont
un jour
ou l’autre
apporté
leur aide avec
la
plus
grande
gentillesse.
Je
garde
unexcellent
souve-nir
des
années que
nous avonspassées
ensemble.
Ces
expériences
n’auraient
pu
voir le jour
sansle travail
des services
techniques
du
Laboratoire :
MM.
THOMME
et
QUILBEUF
pour la
partie
mécanique,
CLERGEAUD,
REGAZZI
et
GYORS
pour
la
partie
électronique,
FLORY
pour
le
remplis-sage des
cellules,
m’ont
chacun
apporté
leur
concours aveccompétence
et
Je
tiens
à
remercier
également
toutes
les personnes
qui
ont
participé
à
la
réalisation
pratique
de
cemémoire
dans
des
conditions
difficiles qui
n’ont
altéré ni
leur
amabilité,
ni
leur talent. Madame MOISSENET a dessiné
une
grande
partie
des
figures,
Monsieur
AUQUIER
aeffectué
de nombreuses
photos.
Le
tirage
de
cetexte
aété assuré
avec unegrande
efficacité
par
Madame
AUDOIN, et
sareliure réalisée dans des délais
très
courts
par
Monsieur
MANCEAU.
Enfin,
Mesdames
BOUNIOL
et
MATYJA
ont
assuré la
frappe
de
cemémoire
avec une
très
grande
compétence ; je
les remercie
pour le
soin
et
la
rapidité
page
PARTIE I - PRESENTATION GENERALE ... 1
I - MOTIVATIONS DE L’ETUDE DE LA MOLECULE
80
Se
2
... 1II - HAMILTONIEN MOLECULAIRE ... 3
1)
Approximation
deBorn-Oppenheimer
... 32)
Hamiltonien et fonctions d’ondeélectroniques : symétries
... 5a) Généralités ...
5b)
Symétries
particulières
des moléculesdiatomiques
homonu-cléaires ... 803B1)
Inversion i ... 803B2) Rotation
C
2
(y)
... 83)
Fonctions d’onde nucléaire -Energies
de vibration-rotation etsymétries ...
94)
Symétries
de la fonction d’onde totale de la molécule -Règles
de sélection ... 13a)
Symétries
... 13b)
Règles
de sélection pourl’opérateur
dipôle
électrique
... 15III - CONFIGURATIONS
ELECTRONIQUES
... 18-
1)
Présentation ... 182)
Configurations électroniques
moléculaires deSe
2
... 193)
Etats moléculaires issus de combinaisons d’étatsatomiques
donnés 21 IV - REVUE DES ETUDESSPECTROSCOPIQUES
DESe
2
... 24PARTIE II - STRUCTURE FINE DES ETATS MOLECULAIRES ... 32
I - GENERALITES ... 32
1)
Rappel
des termesnégligés
... 322)
Termes nondiagonaux
deH
0
...
33a)
Hamiltonien de vibration ... 33page
3)
Calcul d’éléments matriciels ... 354)
Eléments de matrice deH
ND
R
:
casparticulier
desétats
3
03A3
... 37II - HAMILTONIEN DE STRUCTURE FINE RELATIVISTE ... 40
1)
Expression
du Hamiltonien de structure fine ... 40a)
H
SO
... 40b)
H
SR
... 41c) H
OR
...
42d)
H
SS
... 422)
Etudeparticulière
du Hamiltonienspin-orbite
... 43a)
Propriétés
deH
SO
... 43a) Aspect
angulaire ...
4403B2)
Action de i : inversion des électrons ... 4403B3)
Action de 03C3 v ... 45b)
Eléments de matrice deH
SO
... 46a)
Eléments de matricediagonaux
<03C4,~,S,03A3 |
H
SO
|
03C4,~,S,03A3
> 4603B2)
Eléments de matrice nondiagonaux
...
4703B21)
Entre états de mêmemultiplicité
despin
... 4703B22)
Entre états demultiplicités
despin
différentes .... 4803B3)
Symétrisation
des fonctions d’onde de03A9 ~
0 ... 48c)
Eléments de matrice deH
SO
entre états bien décrits par uneconfiguration
électronique ...
493)
Structure fine des états3
03A3
-
... 54a)
Calcul desénergies
et fonctions d’onde d’états3
03A3
-
... 54a)
Structure fine des états3
03A3
... 5403B2)
Transition d’un état3
03A3
-
vers le casc)
de Hund ... 5503B3)
Structureénergétique
des états3
03A3
-
... 57b) Structure fine des états fondamentaux X
3
03A3
-
des molécules du groupe VI-VI ... 60PARTIE III - L’INTERACTION ZEEMAN DES MOLECULES ... 71
I - LE HAMILTONIEN ZEEMAN ... 71
1)
Les différentes contributions àH
Z
... 713)
Propriétés
desymétrie
deH
ZL
... 754)
Eléments de matrice du Hamiltonien Zeeman linéaire dans une base de casa)
de Hund ... 76a)
Eléments de matrice deH’
ZE
... 76b)
Eléments de matrice deH’
ZN
... 77c)
Eléments de matrice deH
ZC
... 775)
Le Hamiltonien Zeemanquadratique
... 78a)
Termediamagnétique
... 78b) Perturbation Zeeman linéaire au second ordre ... 79
c)
Etude dequelques
casparticuliers
... 80c1)
Termes nondiagonaux
en03A9(03A9’ ~ 03A9)
... 80c2)
Termesdiagonaux
en 03A9 :exemple
de l’état3
03A3
-
... 81II - THEORIE PERTURBATIVE DES FACTEURS DE LANDE ... 81
1)
Théorie aupremier
ordre dansl’approximation
deBorn-Oppenheimer
812)
Correctionsélectroniques
au facteurs de Landé ... 833)
Etudeparticulière
du facteur de Landé "de rotation" ... 874)
Facteur de Landé d’un état3
03A3
-
isolé ... 90a)
Traitementperturbatif
... 90b)
Variations deg
J
lors du passage d’un état3
03A3
-
du casa)
au casb)
de Hund ... 925)
Hamiltonien Zeeman effectif dans un état3
03A3
-
... 97PARTIE IV : LES PERTURBATIONS MOLECULAIRES ... 102
I - NOTION DE PERTURBATION ... 102
II - PERTURBATIONS LOINTAINES ... 105
a)
Généralités ... 105b)
Exemples ...
108c) Influence des
perturbations
sur lesspectres optiques :
applica-tion aux transitions B
3
03A3
-
u
~
X3
03A3
-
deSe
2
... 109c1)
Probabilités de transitiondipolaire
électrique -
Durée de vie 109c2)
Transitions3
03A3
-
~
3
03A3
-
... 112c3)
Etudeparticulière
des transitionsperpendiculaires
039403A9 = ±1 .. 114c4)
Observation des transitions croisées3
03A3
1
~
3
03A3
0
:
structure fine des états B et X deSe
2
... 116d) Constantes rotationnelles de l’état B
3
03A3
-
... 123e) Signes
des facteurs de Landé dansl’état
u
-
3
03A3
B
u
deSe
2
... 123III - LES PERTURBATIONS LOCALES ... 124
a)
Etude d’uneperturbation
locale ... 124b)
L’ensemble desperturbations
locales entre deux étatsélec-troniques
donnés ... 1312)
Illustration ... 133a)
Energie
des niveauxperturbés :
constante de rotation effec-tiveB
eff
... 134b)
Variation des facteurs de Landé ... 135c)
Variation des durées de vie03C4
J
... 137d)
Variation de l’intensité relative des transitions croisées B+
u
~ X 1g... 139
e)
Résumé
de ces diverses observations ...
1413)
Etude de l’interaction à 3 niveaux :B 0
+
u
,
B1
u
,
P 1
u
...
142a)
Hamiltonien effectif P1
u
~
B0
+
u
... 142b)
Influence de laperturbation
sur les fonctions d’onde ... 146c)
Diagonalisation
des interactions entre B0
+
u
,
B1
u
,
P
1
u
.. 147c1)
Energies
... 147c2)
Facteurs de Landé ... 150c3)
Transitionsoptiques -
Discussion des intensités ... 153IV - PREDISSOCIATIONS ... 158
1)
Généralités ... 1582)
Prédissociation parcouplage
entre deux états ... 1593)
Prédissociation rotationnelle ... 1604)
Prédissociation au seuil ... 1625)
Nos résultats ... 164PARTIE V - POMPAGE
OPTIQUE
MOLECULAIRE - APPLICATION A L’ETUDE DES FACTEURS DE LANDE DE L’ETAT B3
03A3
-
u
(0
+
)
DE80
Se
2
166 INTRODUCTION ... 166A - THEORIE DU POMPAGE
OPTIQUE
... 167I - INTRODUCTION ... 167
II - FORMALISME GENERAL DE
L’EQUATION
PILOTE ... 1671) Ecriture de
l’équation pilote
... 1672)
Opérateurs
excitation E
et détectionD
... 1703)
Termes de relaxation ... 174a) Le terme de relaxation par émission radiative ... 174
b) Le terme de relaxation par collisions ... 174
c) Le terme de relaxation par
prédissociation
... 175page
III - RESOLUTION DE
L’EQUATION
PILOTE ... 176IV - CALCUL DES SIGNAUX OBTENUS ... 179
1)
Effet Hanle ... 1802)
Résonances en lumière modulée ... 1833)
Battementsquantiques
... 185B - ETUDE EXPERIMENTALE DU POMPAGE
OPTIQUE
DE LA MOLECULE DE SELENIUM ... 188INTRODUCTION ... 188
I - GENERALITES ... 188
1)
Obtention de la vapeur de sélénium ... 1892)
Production duchamp magnétique
... 1913)
Détection de la fluorescence ... 191II - ETUDE DE L’EFFET HANLE ... 193
III - BALAYAGE
MAGNETIQUE
... 195IV - RESONANCES EN LUMIERE MODULEE ... 197
1) Le modulateur
acousto-optique
... 1982)
Détectionsynchrone
de la lumière de fluorescence ... 1993) Résultats ... 199
V - LES BATTEMENTS
QUANTIQUES
ZEEMAN ... 2011)
Laser à colorantpulsé
... 2022)
Détection de la lumière de fluorescence ... 2053)
Résultats : mesure des facteurs de Landé ... 208a)
Facteurs de Landé nonperturbés
... 209b)
Influence desperturbations
... 210c)
Présentation des résultats ... 2104)
Mesures des durées de vie ... 2135) Intensités des transitions croisées B
0
+
~
X 1 ... 215u g PARTIE VI - INTERPRETATION DES RESULTATS OBTENUS ... 216
I - ETUDE DES FACTEURS DE LANDE DANS L’ETAT B
0
+
u
(0 v 6)
DESe
2
... 2161)
Rappel
des 2 modèles utilisés dansl’analyse
de laperturbation
de (B0
+
u
,
v = 4 , J ~ 54) -
Comparaison
... 216 a) Modèle à 2 niveaux ... 216b) Modèle à 3 niveaux (B 0
+
u
, v) , (B 1
+
u
, v) , (P 1
u
, v’) ... 218
c) Justification du modèle à 2 niveaux ... 220
page
a)
Valeurs de A ... 222b)
Valeurs de h ... 224c)
Valeurs deg
B
... 2253)
Résultats obtenus dans l’étude de(B
1
u
,
v = 3)
... 231II - CONSTANTES
SPECTROSCOPIQUES
DE L’ETAT PERTURBATEUR P1
u
... 2321)
"Construction" de l’état P1
u
... 2322)
Détermination des valeurs absolues du nombrequantique
de vibra-tionv(P1)
... 233III - INTENSITE DES TRANSITIONS CROISEES ... 236
IV - DIFFICULTES D’UNE INTERPRETATION COMPLETE DES ELEMENTS DE MATRICE
ELECTRONIQUES
CARACTERISANT L’ETAT B3
03A3
-
u
... 2401)
Interaction B0
+
u
~ B1
+
u
... 2412)
Structure fine de l’étatB
3
03A3
-
u
deSe
2
... 242CONCLUSION ... 244
La connaissance
des
niveauxatomiques
et moléculaires excitésa
longtemps
été limitée parl’élargissement
Doppler
des raiesspectrales.
Depuis
1950,
laspectroscopie
hertzienne des niveauxatomiques
excitéss’est
rapidment développée, grâce
à l’introduction de la méthode de double résonance de Brossel etBitter,
et des diverses méthodes affran-chies de l’effetDoppler
qui ont suivi. Toutes cestechniques
ont permisla détermination d’un très
grand
nombre de structures fines ethyperfines,
facteurs de
Landé,
et durées de vieatomiques.
Ces études n’ont trouvé leur continuation naturelle en
physique
moléculaire que vers l’année 1970. Ceci est à attribuer au manque de
sources lumineuses
capables
d’exciter defaçon
sélective et efficacedes séries de niveaux moléculaires. L’avènement de lasers accordables dans des zones de
plus
enplus
vastes duspectre
lumineux,
et delargeur
spectrale
éventuellement très faible apermis
de dominer cette limita-tion. Il est maintenantpossible
d’étudier par excitation sélective des séries entières de niveauxd’énergie
moléculaires,
et d’effectuersur ces molécules tout un ensemble
d’expériences
despectroscopie
sanslargeur Doppler.
Celles-ci constituent unprolongement
naturelet un
complément
desexpériences
despectroscopie optique.
Nous avons décidé de nous intéresser à la molécule
diatomique
de Sélénium. Cette
molécule,
de même que toutes celles de la famille del’oxygène, présente
des étatsélectroniques paramagnétiques
(S =1);
on
peut
deplus
en obtenir desisotopes
purs sans spin nucléaire. Elle a, sur les moléculesplus légères
(oxygène
etsoufre),
l’avantage
d’absorber dans l’ultraviolet
proche
(03BB ~
4200A),
zone où existaientdés le début de ce travail des lasers à colorant aisément utilisables.
Par
ailleurs,
les travaux del’équipe
du Professeur BARROW avaientdéjà
établi les bases d’une bonne connaissancespectroscopique
de l’état B3
03A3
-
u
que nous avons étudié.Ce mémoire est
composé
de 6parties :
les troispremières
La
partie
Irappelle
les bases del’approximation
deBorn-Oppenheimer
et fait lepoint
sur l’état des connaissances concernant laspectroscopie
de la molécule de Sélénium.
Compte
tenu de lagrande
importance
de seseffets,
une étude del’interaction
Spin-Orbite
estmenée
dans lapartie II,
etappliquée
aux états
moléculaires
3
03A3
auxquels
nous nous sommes interessés.La
partie
III est consacrée au Hamiltonien Zeeman desmolécules,
à sessymétries
et à la théorie du calcul des facteurs de Landé.La
partie
IV aborde l’étude desperturbations
moléculaires etde leurs diverses manifestations. Nous y
présentons
enparticulier
une étude très détaillée d’une
perturbation
locale de l’état(B
0
+
u
,
v = 4)
, observée à travers unegrande
variété dephénomènes.
La
partie
V est consacrée à l’étude du pompageoptique
molécu-laire. Dans unpremier
temps
nousrappelons
la théorie desexpériences
d’effet
Hanle,
de Résonance en Lumièremodulée,
et de BattementsQuantiques,
en insistant sur les liens unissant ces différentsphéno-mènes. Nous
présentons
dans un secondtemps
l’ensemble de nos résultatsexpérimentaux
qui concernent les facteurs deLandé,
durées de vie, etspectres
de fluorescence de l’état B3
03A3
-
u
du Sélénium.Ces résultats sont
interprétés
dans lapartie
VI,
etpermettent
de décrire très finement les
perturbations
induites dans cet état molé-culaire par l’interaction avec l’étatperturbateur
3
03A0
1 .u
Nous
présentons
dasn unappendice
l’ensemble des méthodesnumé-riques de calculs moléculaires que nous avons
utilisées,
et unePARTIE I
PRESENTATION GENERALE
I - MOTIVATIONS DE L’ETUDE DE LA MOLECULE
80
Se
2
L’intérêt de notre
équipe
s’estporté
sur l’étude du pompageoptique
desmolécules,
enparticulier
par la mesure de diversespropriétés
d’états excités des moléculesdiatomiques :
facteurs deLandé,
durées devie,
struc-tures
hyperfines...
Après
des travaux sur NO[
1
]
et le début des travaux surI
2
[
2,3
],
nous avons dû choisir un sujet
d’étude,
cequi
est relativementmalaisé,
compte
tenu du nombre de molécules existantes et de la diversité de leurspropriétés.
Deuxoptions
sont, enparticulier, possibles :
choisir une molé-cule demanipulation
aisée,
et absorbant dans lespectre visible,
motivationsqui
ont fait le succès des travaux surl’Iode,
ou bien essayer d’étudier unemolécule
plus légère
et d’intérêtthéorique
plus grand,
comme parexemple
CO[
4
],
les difficultésexpérimentales
étant alors considérables.Notre choix de la molécule de Sélénium
(Se
2
)
est unhybride
de cesdeux tendances. D’un
point
de vuethéorique,
nous cherchions une moléculesans structure
hyperfine
(I = 0),
contrairement àl’Iode,
etparamagnétique,
l’Oxygène O
2
(molécules
du groupeVI-VI).
Parcontre,
il fallait une molécule d’étudespectroscopiquement
aisée : les bandesd’absorption
deO
2
se trouvent dans l’ultraviolet lointain(03BB
< 2000Å)
etsont,
de cefait,
difficilement accessibles par excitationlaser ;
deplus,
l’état excitécorrespondant
est fortementprédissocié,
cequi
n’ensimplifie
pas l’étude.Cependant,
quand
la masse moléculaire
augmente
dans la famille, S
2
O
2
, Se
2
,
Te
2
,
la zoned’absorption
sedéplace
vers levisible,
commel’indique
le tableau suivant.Zones
d’absorption
des molécules du groupe VI-VI homonucléaires(transitions
X3
03A3
-
g
~
B3
03A3
-
u
, v"=0 , v’=0)
g u
La molécule
S
2
absorbe dansl’ultraviolet,
dans une zone où il existaitpeu de lasers accordables au moment du début de ce travail. La molécule
Te
2
absorbe dans lebleu,
mais, en raison de sa masseimportante,
son spectred’absorption
est très dense. Dans cettefamille,
nous nous sommes arrêtés àSe
2
qui
représentait
uncompromis acceptable.
Des différentsisotopes
de34
Se
(74
A82),
leplus abondant
est80
Se
(50%
dumélange
naturel)
; il peut
être obtenu avec une
pureté
supérieure
à 98% pour un coût modeste. Lapression
de vapeur de’Sélénium est de 1 Torr à unetempérature
de 350°C[
5
] ;
un four deconstruction
simple permet
donc d’obtenir cette vapeur.Par
ailleurs,
un certain nombre de travaux[
6,7,8
]
avaient donné uneconnaissance
spectroscopique
assez bonne des transitionsX
3
03A3
-g
~ B
3
03A3
-u
deSe
2
et aussi montré l’intérêt d’une excitation laser sélective de cette molécule. Nouspensions
donc être bien armés pour entamer des étudesplus
fines de l’étatB
3
03A3
-
u
.
Par ailleurs,
le travail deDalby,’au
laboratoire[
9
]
arapidement
donné de
premiers
résultats,
tels que l’observation’de l’effet Hanle surAvant d’aller
plus
loin dans l’étudeparticulière
de la moléculeSe
2
nous allons
rappeler
un certain nombre depropriétés
et denotations
molëcu-laires,
en lesexplicitant,
biensûr,
sur cetexemple
concret.II - HAMILTONIEN MOLECULAIRE
1)
Approximation
deBorn-Oppenheimer
Dans un
premier
temps,
nousconsidérons
uniquement
lesinteractions
électrostatiques
entre électrons et noyaux, pour une molécule isolée. Comme il estclassique
(par
ex. Judd[
10
]),
nous étudions le mouvement d’une molé-culed’impulsion
totale nulleP=03A3p
t
+p
A
+p
B
=0 ,
(l’indice
ts’appliquant
aux
électrons,
A et B aux 2noyaux)
endistinguant
les variables concernant le mouvement relatif des noyaux(|r
A
-r
B
=r
BA
r
BA
|
= r),
et les variables concernant lemouvement
desélectrons,
rapportées
au centre de masse C des noyauxLes
impulsions
associées àr
BA
etr
tc
sontrespectivement
p
BA
etp
tc
.
Le Hamiltonien totalH
0
de lamolécule,
dans le référentiel du centre de masseet en fonction de ces
coordonnées,
s’écrit :03BC est la masse réduite des 2 noyaux 03BC:
Le choix des coordonnées que nous avons effectué
permet
unequasi-séparation
desénergies
cinétiques
nucléaire etélectronique :
seul le 3ème termeA
+M
B
)]
-1
[2(M
(03A3
2
, a
p
)
tc
priori
petit,
traduit l’effet de recul des noyaux, bien connu dans les atomes. Ce terme de recul estplus petit
que le termed’énergie cinétique
électronique,
dans unrapport
d’environm/M
A
+M
B
.
Nous pouvons scinder
H
0
en 2 termes, l’unélectronique,
l’autre
nu-cléaire : le Hamiltonienélectronique
H
E
s’écrit : et leHamiltonien
nucléaire :
Endécomposant
p
BA
suivant sescomposantes,
parallèle
etperpendiculaire
à l’axeinternucléaire,
onpeut
écrire :le
premier
de ces termesreprésentant
l’énergie
cinétique
devibration
molé-culaire(oscillation de
la distanceinternucléaire
r),
et le deuxième termel’énergie cinétique
de rotationmoléculaire.
Une
comparaison
[
11
]
des ordres degrandeurs
desfréquences
propres desmouvements
électroniques,
de vibration,
derotation,
montre que cesfréquences
sont
proportionnelles à 1
, (m 03BC)
½
,(m 03BC) .
Dans ces
copditions,
les vitessesdes électrons
sont trèslargement
supérieures
aux vitesses de mouvement des noyaux. Autrementdit,
l’approxima-tion de
Born-Oppenheimer
[
11
]
admet que les électrons suiventadiabatiquement
le mouvement des noyaux.Réciproquement,
lepotentiel
"vu" par les noyaux résulte d’une moyenne sur le mouvement des électrons.Il est alors naturel de
chercher
desfonctions d’onde
moléculaires factorisées :La fonction
d’onde 03C8
N
nedépend
que des variables orbitales nucléairesr=
|r
BA
|
et 03C9~(03B8,
~)
caractérisant l’orientation de l’axe internucléaire dans le référentiel du laboratoire. Parailleurs,
la fonction d’ondeélectro-nique
03C8
E
ne
dépend
alors de r que comme d’unparamètre. Enfin,
03C8
I
est
la
fonction d’onde
despins nucléaires,
ceux-ci n’intervenant que par deseffets
de
symétrie.
Nousadopterons
comme base de fonctions d’onde desfonctions
dece
type ;
les termes du Hamiltonien total moléculaire violantl’approximation
de
Born-Oppenheimer
apparaitront
comme depetits
termes nondiagonaux
dans cette base.2)
Hamiltonien et fonctions d’ondeélectroniques :
symétries
a)
GénéralitésOn
peut
diagonaliser
leHamiltonien
électronique
H
E
pourchaque
valeur de la distance internucléaire r . On obtient ainsi les étatsélectroniques
03C8
e
(r
tc
,
r)
d’énergie
E
e
(r) :
(l’ensemble
des étatsélectroniques
à r fixé forme biensûr
une base des fonctions d’ondeélectroniques).
Soit L le moment
cinétique
orbital desélectrons
(calculé
parexemple,
par
rapport
à C :L
=03A3
r
tc
~
p
tc
) ;
L ne commute pas avec le Hamiltonienélectronique H
E
,
mais laprojection
L
z
deL
sur l’axe internucléaireAB=Oz
commute avec
H
E
,
à
cause de lasymétrie
cylindrique :
Par
conséquent,
si L
2
n’est pasquantifié,
Lz l’est et
on caractérise les dif-férents états
électroniques
par la valeur du module de Lz
/h
notée A .La notation
spectroscopique classique
est que suivant la valeur deLe Hamiltonien
H
E
commute aussi avecl’opérateur
desymétrie
dans unplan
passant
par l’axeinternucléaire
AB , noté
03C3
V
.
Par contre, les deux
opérateurs
L
z
et03C3
V
ne commutent pas ;ils
anticommutent :Il en résulte que :
i)
l’état obtenupar
action de03C3
V
sur l’état|~>
est un état propre deL
z
,
de
valeur propre-~,
ii)
ces 2 états sontdégénérés
si~ ~ 0 ,
iii)
les états de A = 0 sont états propres de03C3
V
,
de valeurs
propres +1 ou-1 ;
ces états sontnotés respectivement
03A3±(E
car~=0)
Le
Hamiltonien H
E
ne tient pascompte
desinteractions
relativistes liéesaux
spins
électroniques
(termes de structurefine) :
toutefois,
lespin
total S =03A3 s
t
intervient dans la caractérisation d’un étatélectronique,
par l’in-termédiaire duprincipe
de Pauli.A
l’évidence,
S2 et lacomposante
de S sur un axequelconque,
enpar-ticulier
S
z
commutent avecH
E
:
On note S(S+
1)
la valeur propre deS
2
/h
2
et 03A3 la valeur
propre deS
z
/h .
Lanotation
spectroscopique
complète
d’un étatélectronique
est :Le nombre
quantique 03A9
est laprojection
du momentcinétique électronique
total sur l’axe internucléaire :Nous suivons ici la notation de
Hougen
[
12
] : 03A9
est déterminé en valeuralgébrique
(03A9
estnégatif
si 03A3 < 0 et|03A3| >
A).
Nousadoptons
donc pour caractériser la fonction d’ondeélectronique,
la notation deket | T ,
A ,
S , 03A3 , 03A9
>(
représentant
lesparamètres
annexes del’état).
Une telle basecorrespond
au cas decouplage
de Hund a.Avant d’aller
plus
loin,
il fautpréciser
l’action de03C3
V
sur lapartie
despin
de la fonction d’ondeélectronique.
03C3
V
commute avecS
2
, mais
pas avecS
z
.
Si l’on choisit les conventions dephase
usuelles pour les fonctionsd’onde
[
13
], l’action
de03C3
V
sur la fonction d’onde despin |
S , 03A3 >s’écrit :
Etudions par
exemple,
l’action de03C3
V
sur un état3
03A3
-
(S = 1
, ~=0),
on voit aisément que ,donc
La
première
de ceséquations
montre que l’état 03A9=0 issu d’unétat
3
03A3
-
estpair
vis-à-vis de03C3
V
,
la
deuxièmeéquation
montresimplement
que les états 03A9=±1 se transforment l’un dans l’autre par03C3
V
.
Notons d’ailleurs que le Hamiltonien
électronique
total,
incluant tousles effets relativistes
(mais
avectoujours
les noyaux immobiles) commutetoujours
avecL
z
+S
z
(maisplus
avecL
z
ou
S
z
séparément),
car l’axe inter-nucléaire reste un axe desymétrie
de révolution :03A9 reste donc un bon nombre
quantique,
même si A et 03A3 ne le sontplus.
03C3
V
commute lui aussi avec cette forme
plus générale
de H : E et03C3
V
anticommuteavec L
z
+S
z
:
Les
propriétés
énoncéesplus
haut avecL
z
et ~
restent exactes si on les énonce avecL
z
+
S
z
et 03A9
; enparticulier,
un état de 03A9=0 atoujours
uneparité
définie vis-à-vis de03C3
V
:
b)
Symétries particulières des molécules diatomiques homonucléaires
03B1)
Inversion iPour une molécule
ayant
deux noyaux de mêmecharge,
l’inversion i descoordonnées des électrons par
rapport
au centre decharge
des noyaux est uneopération
desymétrie :
On
distingue
les fonctions d’onde à caractère g ou u (en allemandgerade
etungerade),
telles que :Si de
plus
les noyaux sontidentiques
(mêmecharge
et aussi mêmemasse),
onmontre
[
2
]
que i commute avec le Hamiltonien moléculairetotal,
àl’exception
des termes
hyperfins
[
2
]
et de certains termes decouplage
avec deschamps
extérieurs. Afortiori,
en l’absence despins
nucléaires,
le Hamiltonientotal d’une molécule isolée commute avec i .
03B2)
RotationC
2
(y)
Toujours
pour une molécule ayant deux noyaux de mêmecharge,
larota-tion de 03C0 des électrons autour d’un axe
perpendiculaire
à l’axe internucléaireet
passant
par le centre decharge
est uneopération
desymétrie
pour la fonc-tion d’ondeélectronique,
notéeC
2
(y) :
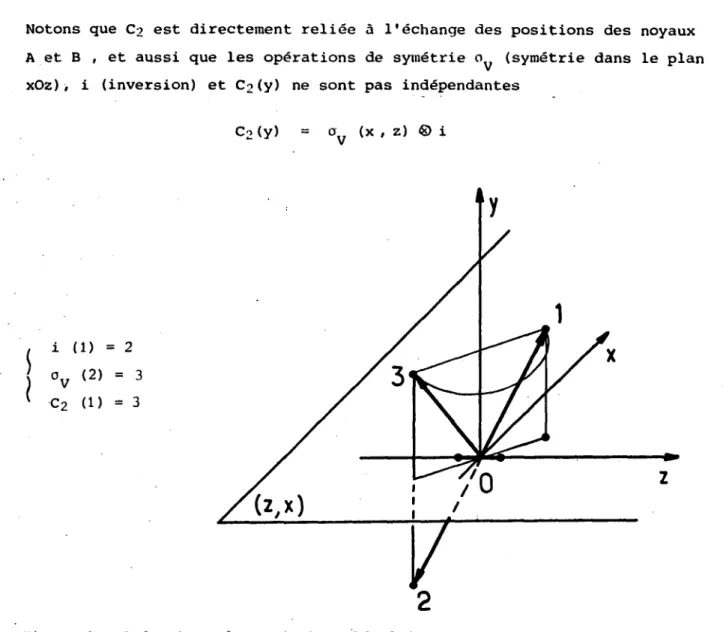
Notons que
C
2
est directement reliée àl’échange
despositions
des noyaux A et B , et aussi que lesopérations
desymétrie
03C3
V
(symétrie
dans leplan
xOz),
i(inversion)
etC
2
(y)
ne sont pasindépendantes
Figure
1 :Opérations
desymétrie
moléculaires.3) Fonction d’onde nucléaire -
Energies
de vibration-rotation etsymétries
Nous avons vu que le mouvement nucléaire de la molécule est une
compo-sition de vibration et de
rotation ;
on cherche donc la fonction d’ondenu-cléaire orbitale sous forme du
produit :
où
03A6
R
(03C9)
est la fonction d’onde derotation,
et~
v
(r)
la fonction d’onde de vibration. (Le facteur1/r provient
de ce que r est une variable radiale).La
fonction 03C8
N
est, dansl’approximation
deBorn-Oppenheimer,
solution de :E
E
(r)
estl’énergie électronique
de l’état moléculaire considéré pour lavaleur r de la distance internucléaire.
L’expression
deH
N
permet
dedécou-pler
la rotation et la vibration :En l’absence de rotation
(E
R
=0)
lepotentiel
de l’étatélectronique
considéréU(r)
est doncégal
àE
E
(r) ,
et la fonction d’onde de vibration est solution del’équation
deSchrédmger
usuelle :Notons que cette fonction
qui
nedépend
que de r =|r
AB
|
n’est
pas affectée par les diversesopérations
desymétrie
de la molécule. Si lepotentiel
molé-culaire U(r)présente
un minimum suffisammentimportant,
il y aura des états discrets de vibration notés|v>
et dans tous les cas un continuum d’états pour E U(~) .L’énergie
moléculaire de vibration est celle d’un oscillateuranharmonique
etl’énergie
de l’état|v>
s’écrit :v est un entier
positif
ounul,
T
e
désigne
l’énergie
du minimum dupotentiel
U(r) . (Comme
il est usuel enphysique
moléculaire,
nousexprimerons
lesénergies
et les divers coefficientsspectroscopiques
en unité usuelle de nombred’onde,
c’est-à-dire encm
-1
).
La fonction d’onde de
rotation 03A6
R
(03C9)
est fonction propre du Hamiltonien derotation
H
R
:
Le moment
cinétique
de rotation des noyaux R estégal
au momentcinétique
total J de la molécule (sans spins nucléaires) diminué des moments
cinétiques
Pour une molécule isolée sans
spins
nucléaires,
J et M
J
sont de bons nombresquantiques ;
si deplus,
03A9=~+03A3 est défini(cas
de Hund a ouc),
la fonction d’onde de rotation est caractérisée par les 3 nombres
quantiques
J ,
M
J
,
03A9 . Nous noterons le ketcorrespondant |
J ,
03A9 ,
M
J
> .Le choix de normalisation et de
phase
leplus
couramment utilisé[
12
]
pour décrire la fonction d’onde de rotation est :D
J
M03A9
étant
la matrice de rotation de l’axe internucléaire dans la direction03C9 =
(03B8 ,
~) .
Le terme
E
R
introduitplus
haut est lapartie
de
H
R
diagonale
en étatélectronique
et se calcule comme suit :où nous avons omis certains
petits
termesindépendants
de J et que l’on sup-poseintégrés
àU(r) ;
les termes nondiagonaux
en étatélectronique
serontconsidérés à la fin de ce
paragraphe.
SiJ(J+ 1)
n’est pas trèsgrand,
onpeut
calculersimplement
l’effet deE
R
enperturbation
aupremier
ordre etl’énergie
totale de l’état s’écrit :où
B
v
=
<v|
2
203BCr
2
|v>
est la constante de rotation de l’état v . Cettecons-tante de rotation
dépend
de v et on l’écrit leplus
souvent comme unpoly-nome en
(v+½)
.et on montre aisément que
B
v
s’annule à la limite de dissociation. Si J(J+ 1)est assez
important,
il faut calculer de manièreplus
précise
lesénergies ;
effets de distorsion
centrifuge
de la molécule. Onreprésente
de manièresystématique
ces effets en écrivant lesénergies
sous la forme due à Dunham[
14
] :
Les coefficients de Dunham
Y
ij
sont directementreliés,
aux termes du 2èmeordre en
(B
e
/03C9
e
)
près,
aux coefficientsspectroscopiques précédemment
définis:Dans cette
approximation,
Y
ij
estproportionnel
à(03BC)
-(j+i 2)
.
Cette relationest très utile pour comparer des molécules de formules
isotopiques
différentes. Si 03BC1 et 03BC2 sont les masses réduites de deux moléculesisotopiques,
lescoef-ficients de Dunham sont liés
par
la relationT
e
étant le mêmepour les
deuxisotopes.
(Ceci
suppose que lepotentiel
U(r) =E
E
(r)
estindépendant
del’isotope ;
cela revient ànégliger
totalement l’effet dupetit
terme deH
E
:
1 2
(M
A
+M
B
)
(
03A3
p
tc
)
2
).
Par
ailleurs,
le coefficientY
00
dudéveloppement
de Dunham n’est pas nul engénéral
si lepotentiel
U(r)
n’est pasharmonique
[
14
].
On montre que :L’énergie
exacteT
e
du fond dupuits
depotentiel
est obtenue enextrapolant
E(v ,
J) àJ(J
+
1)
= 03A9
2
et àv
min
= -1 2 -
0394 avec 0394 =Y
00
/03C9
e
.
Cette différence 0394 , relativement faible, n’est pasnégligeable
compte
tenu de laprécision
des mesuresspectroscopiques ;
on devra en tenircompte
enparticulier
dans la reconstruction dite R.K.R. dupotentiel
moléculaire àpartir
des donnéesspectroscopiques.
Revenons au Hamiltonien de rotation
H
R
;
dans
labase |
03C4,~,S,03A3,03A9,J,M
>(Ox et
Oy
étant 2 axes liés à la molécule dans leplan perpendiculaire
à l’axeinternucléaire).
D’après
lespropriétés
desopérateurs
vectoriels,
il est clair queH
ND
R
couple
des états de 03A9 différents de uneunité ;
il viole doncl’approxi-mation de
Born-Oppenheimer.
Celle-ci n’est donc valable que pour des états de momentcinétique
de rotation faible ainsiqu’on pouvait s’y
attendre.4)
Symétries
de la fonction d’onde totale de la molécule -Règles
de sélec-tionoptiques
a)
Symétries
Nous avons rencontré 3 transformations de
symétrie,
s’appliquant
auxmolécules
diatomiques
symétriques :
03C3
V
,
i ,
C
2
.
Cesopérations
sont définies dans le référentiel moléculaire et laissent donc celui-ci intact. Simainte-nant on considère l’ensemble de la
molécule,
électrons et noyaux, dans leréférentiel du
laboratoire,
on peut définir 2 nouvellesopérations
desymétrie :
- l’inversion I de toutes les
particules
parrapport
au centre de masse,- si la molécule est
homonucléaire,
l’échange
P des 2 noyaux.Ces deux
opérations
sont évidemment des transformations desymétrie
du Hamiltonien total :Soit 03C8
la fonction d’onde totale de la molécule :où 03C8
E
, 03C8
N
,
03C8
I
sontrespectivement
les fonctions d’ondeélectronique,
nucléaire
orbitale,
et despin
nucléaire.Si la fonction
d’onde 03C8
est convenablementsymétrisée,
elle se trans-forme en elle-même ou en sonopposée,
par action de I ou P .Si
I|03C8> = ±
|03C8>
le niveau estrespectivement
deparité
(+)
ou(-).
Si
P|03C8> = ±
|03C8>
le niveau estsymétrique
(s) ouantisymétrique
(a).
Etudions d’abord
l’opération
d’inversion I : nous considéronsqu’elle
porte
sur les coordonnées deposition
et despin
des électrons et des noyaux :une
analyse
détaillée[
10
]
montre que l’action de 1 estéquivalente
à celle de03C3
V
,
définie
dans le référentielmoléculaire,
maisagissant
à la fois surles fonctions d’ondes
électronique
et de rotation.Ainsi, reprenant
l’exemple
des états3
03A3
-
:
Soit, si 03A3 = 03A9 = 0 :
Et
pour 03A9 ~ 0 ,
en prenant les combinaisons de +03A9 et -03A9 :Si l’on dénote
par |
03A9=1
+
>
les combinaisons obtenues avec lessignes
+ et-on trouve :
Les niveaux rotationnels des états 03A9=0 et
03A9=1
+
ont donc laparité
(-1)
J
,
et ceux de l’état 03A9=
1
-
laparité
.
(-1)
J+1
Onpeut
de même étudier laparité
des niveaux rotationnels d’un état~~0 .
Considérons maintenant l’action de la
permutation
P des noyaux. A cetégard,
le cas de80
Se
2
estparticulièrement
simple,car
lesspins
nucléaires sont nuls(I
1
= I
2
= 0) ;
les noyaux sont donc des bosons. La fonction d’onde de spinnucléaire 03C8
I
est l’unité, et Pn’aqit
quesur 03C8
N
.
D’autre part,l’échange
de 2 bosons doit laisser la fonction d’ondeinchangée :
tous les niveaux de80
Se
2
sont doncsymétriques (s).
En l’absence de
spins
nucléaires,
l’opération d’échange
P estsimplement
la
composition
de l’inversion de toutes lesparticules
I , par l’inversion des électrons seuls iLes actions de I et i
ayant
déjà
étéétudiées,
on endéduit,
pour des états3
03A3
,
les résultats suivants :Tous les niveaux devant être
symétriques,
il suit que pour les états :2022
3
03A3
-
g
(
, 0
+
g
etg
)
+
1
et(
3
03A3
-
u
, 1
-
u
),
seuls existent les niveaux de Jpair.
2022
03A3
(
3
-u, 0
+
u
et1
+
u
)
et-
g
3
03A3
(
,
1
-
g
),
seuls existent les niveaux de Jimpair.
En
conséquence,
tous les niveaux g considérés(
3
03A3
-
,
+
g
, 03A9=0
1
+
g
,
1
-
g
)
sont
pairs
(+),
et tous les niveaux u sontimpairs
(-).
Ces différents ré-sultats sont rassemblés dans le tableau suivant :b)
Règles
de sélections pourl’opérateur dipôle électrique
Pour une molécule à noyaux
identiques
(comme80
Se
2
),
ledipôle
D est un
opérateur
vectorielorbital ;
on en déduit lesrègles
de sélectionsur la fonction d’onde de rotation :
et sur la fonction d’onde
électronique :
D
z
commute avec03C3
v
;
comme c’est ce terniequi couple
les états de même valeur de A oude 03A9 ,
on en déduit que :On vérifie que D anticommute avec les inversions i
(des électrons)
et I(de
toutes les
particules).
Seules sont autorisées les transitions u ~ g et entre états de
parité
totaleopposée :
Enfin,
D commute avecl’échange
des noyauxidentiques ;
les transitions auto-risées conservent cettesymétrie :
Pour les transitions B
3
03A3
-
u
~ X3
03A3
-g
de80
Se
2
,
celles-ci segroupent
donc en 2
sous-systèmes
correspondant
à039403A3 = 0 ,
0
+
g
~g
0
+
et1
±
g
~
1
±
u
:
2022 Système
0
+
g
~0
+
u
:
J
g
estpair
etJ
u
impair.
Il existeuniquement
des raies P et R :Système
Les transitions
1
+
g
~1
-
u
etg
1
-
~1
+
u
sont très faibles(raies Q , 039403A9=0).
2022 Les transitions 1
±
~
0
+
et
1sont
±
+
~ 0
interdites tant que S et 03A3 sont g u u gbien définis.