HAL Id: hal-01180136
https://hal.archives-ouvertes.fr/hal-01180136
Submitted on 27 Jul 2015
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
La Matière organique dans les milieux naturels : actes
des neuvièmes journées du Diplôme d’Etudes
Approfondies Sciences et techniques de l’environnement,
organisées les 14 et 15 mai 1998, à Paris
Christian Le Coz, Bruno Tassin, Daniel Thevenot
To cite this version:
Christian Le Coz, Bruno Tassin, Daniel Thevenot.
La Matière organique dans les milieux
na-turels : actes des neuvièmes journées du Diplôme d’Etudes Approfondies Sciences et techniques de
l’environnement, organisées les 14 et 15 mai 1998, à Paris. Journées du Diplôme d’Etudes
Appro-fondies Sciences et techniques de l’environnement, May 1998, Paris, France. Presses de l’Ecole
na-tionale des ponts et chaussées, pp.147, 1998, Actes des Journées du Diplôme d’Etudes Approfondies
en Sciences et techniques de l’environnement, 978-2-85978-319-8. �hal-01180136�
Sous la direction de Christian LE
COZ,
Bruno TASSIN
etDaniel
THÉVENOT
LA
MATIERE
ORGANIQUE
DANS LES
MILIEUX
NATURELS
Actesdes neuvièmesjournées duDiplômed'ÉtudesApprofondies
Sciencesettechniquesde l'environnement
organisées les 14et15mai 1998, à Paris
UNIVERSITÉ FACULTE— PARIS cte
VAL de
MARNE SCIENCESETTECHNOLOGIE
D £ÉS
ENGREF
Chez lemême éditeur
Pluieetenvironnement(1998)
sousla direction de Ch. LeCoz, B. TassinetD. Thévenot
Mesuresetenvironnement(1997)
sousla direction de Ch. LeCOZ, B. TASSINetD. THÉVENOT
Transfert despolluants dans leshydrosystèmes (1996)
sous la directionde,Ch. Le Coz, B. TassinetD. Thévenot
L'eaudans la ville(1995)
sousla directionde C. LELONGetJ.-C. DEUTSCH Gestionintégrée des milieux aquatiques (1994)
sousla directionde Ch. Le Coz
Lavilleetlegénie de l'environnement (1993) sousla direction de B. BARRAQUÉ
Rejets urbainspartempsdepluie: pollutionsetnuisances (1993)
sousla directiondeB. TASSINetD. THÉVENOT
Gestion deseaux(4 vol.)
sousla direction de F. VALIRON
DANGER
PHOTOCOPILLAGE TUE LE LIVRE .
©1999
Le code de la propriété intellectuelle du 1" juillet 1992 interdit expressément la
photocopieàusagecollectifsansautorisation desayantsdroit. Or,cettepratiques'est
généralisée,notammentdansl'enseignement,provoquantunebaisse brutale desachats
de livres, au point que la possibilité même pour les auteurs de créer des œuvres
nouvellesetde lesfaire éditercorrectement estaujourd'huimenacée.
Nousrappelons doncquetoutereproduction, partielleou totale, duprésentouvrage,
sur quelque support que ce soit, est interditesans autorisation de l'auteur, de son
éditeur ou du Centre français d'exploitation du droit de copie (CFC, 20 rue des
Grands-Augustins,75006 Paris).
ISBN2-85978-319-9
1*6 S S 0 Sde l'école nationale dos
onts et
chaussées
28,ruedes Saints-Pères
Sommaire
Caractérisationde lamatièreorganique
Caractérisationetévolution desorganohalogènes dans l'eau demer 7
Anne-Sophie Allonier, Valérie Camel, Alain Bermond,
Michel Khalanski
Oxydation de molécules organiques dissoutesau coursdes traitements del'eau :
rôle des réactionsradicalaires 19
AnneVentura,Valérie Camel, Alain Bermond
Évolutionde lamatièreorganique dans les milieux naturels
Dégradation dela matière organiqueetprocessusbiogéochimiques
misenjeu dansuneberge de l'Essonne 33
Cyril Moneron, Jean-MarieMouchel
Biodégradabilité de la matière organique dans les systèmes aquatiques
urbains : l'exemple de l'agglomération parisienne 49
Pierre Servais
Impact de la dégradation de lamatière organique surles teneursenoxygène dissous
dansuncours d'eauurbanisé 63
MarcDelbec,Jean-Marie Mouchel
Coagulation of colloidalmaterial in surfacewaters: the roleof natural
organicmatter 81
K.J.Wilkinson, J.-C. Nègre,J. Buffle
Interactionentrelamatièreorganiqueetles micropolluants
Différentsaspectsdes interactionsentreles matières organiques des sols
etlesmicropolluants minéraux 93
Philippe Cambier, Isabelle Lamy
Matièreorganiquedes solsetmicropolluants organiques 107
E.Barriuso, P. Benoit, S. Houot, M.P. Charnay
Approche expérimentaleetpar modélisation de la complexation du cuivre
parles matières organiques naturelles 119
Rayna Charlatchka, Philippe Cifroy
Remobilisation parl'EDTA du cadmiumetdu plomb accumulés dansun sol
et unsédiment de rivière contaminée 139
CARACTERISATION ET EVOLUTION DES ORGANOHALOGENES DANS L'EAU DE MER
Anne-Sophie
ALLONffiR1'2,
ValérieCAMEL2,
AlainBERMOND2,
MichelKHALANSKI1
1EDFDirectiondes EtudesetRecherches,Département Environnement
BP49, 78401 CHATOU Cédex,France
2
INAP-G, Laboratoire de ChimieAnalytique, 16rue Claude Bernard75005Paris
Résumé
Dans les centrales nucléaires implantées sur le littoral marin, l'eau de mer est utilisée comme
fluide réfrigérant. Cette eaucontientdes larves de multiples espèces d'invertébrésmarinsquise
fixentsurlesparoisdescircuits.Afin de garantir ladisponibilité delasourcefroide, ilconvient
d'éviter ledéveloppement excessif de ces organismesfixés surlesparois des canalisationsdans
lesquelles circule l'eau de mer. Diverses méthodes ont été développéespour lutter contre les
salissuresbiologiques. Le traitement chimique le plus couramment utilisé dans les circuits de
refroidissementalimentésen eaudemerestla chloration.
L'addition dechlore dans l'eau de merconduit, par des réactions complexes, à la formation de composéshalogénés. Lesproduits oxydants généréssontprincipalement de l'acide hypobromeux
et, enprésenced'azote ammoniacal, des bromamines : ils disparaissent rapidement après rejet
dans le milieu récepteur. D'autres sous-produits sont en revanche plus persistants. Il s'agit
d'organohalogénés majoritairementbromés. Parmi ceux-ci, lescomposés volatils sontdosables
parchromatographicenphasegazeuse(CPG) après extractionpar«purgeandtrap» ;ils sont
constitués en grande majorité par le bromoforme. Les composés moins volatils comme les
haloacétonitrilesetles halophénolspeuventégalementêtredosésparCPG après extractiondans
unsolvant. Nous avonsdéterminéexpérimentalement la cinétique de formationet le rendement deformation du bromoforme, du dibromoacétonitrile et du 2,4,6-tribromophénol dans les eaux demerprovenantde diverssitesde centralesnucléaires, chloréesaulaboratoire.Lebromoforme
est rapidement généré, dansune gamme de chlorations allant de 0,5à 4,0mg/len chlore, avec
un rendementmassiqueproche de3%.Ledibromoacétonitrileestplus lentement généréavec un rendementmassiquede l'ordre de0,3%. Le rendementmassiquedu 2,4,6-tribromophénol esttrès
faible (0,03%) pour une gamme de chlorations allant de 0,3 à 10 mg/l.
Des mesures par CPG effectuées sur les eauxde rejetprovenant de divers sites de centrales nucléaires qui pratiquent la chloration à bas niveau de leurs eaux de refroidissement (Gravelines, Penly et Paluel sur le littoral de la Manche) confirment ces résultats sur le
bromoforme et indiquent également la présence de dibromoacétonitrile et de 2,4,6-tribromophénol, à plus faibleconcentrationquele bromoforme.
1. Contexte
Dans les centrales nucléaires implantées sur le littoral marin, l'eau de mer est utilisée comme fluide réfrigérant. Chaque tranche nucléaire prélève ainsi un débit d'eau de 40 à 45
m3/s
; cette eau contient des larves de multiples espèces d'invertébrés marins (mollusques, bryosoaires,hydraires...)qui sefixentsurles parois des circuits. Afin de garantir ladisponibilité de lasource
froide, il convient d'éviter le développement excessif d'organismes fixés sur les parois des
canalisations danslesquelles circule l'eau demer.Ces organismes indésirables,queles industriels
appellent «salissures biologiques»peuvent, eneffet, nuire au bon fonctionnement des centrales
et, lorsqu'ilsse détachent, ils peuventcolmater les échangeurs thermiques. Il est donc nécessaire
de contrôlerceprocessusdans leseauxutiliséespourlescircuits de refroidissement.
Diverses méthodes sont mises en œuvre pour lutter contre les salissures biologiques (Morizur,
1985) notammentle nettoyage mécaniqueen continu des tubes de condenseurpardes boules en caoutchouc qui élimine en permanence les dépôts organiques, l'utilisation de peinture anti¬
salissures, les chocs thermiques et salins (passages successifs d'eau de mer à 45°C et d'eau
douce). Toutefois la chlorationest, depuis longtemps, et resteencore aujourd'hui, le procédé le plus couramment utilisé dans le monde. La chloration continue à faible dose, du printemps à
l'automne,aainsi étéadoptéeparEDFpourtraiter l'eau derefroidissement des centrales à flamme
ounucléairessituéesenbord demer.Ceprocédé offre desavantages surle plan de l'efficacité, du
coûtet de l'acceptabilité environnementale par rapport à d'autres traitements chimiques. L'agent
chloré utilisé est l'hypochlorite de sodium, produit sur le site même par électrolyse de l'eau de
mer.
2. Chimie du chlore
L'addition de chlore dans l'eau de refroidissement conduit, par des réactions complexes, à la formation de composés halogénés. Bien que la plupart de ces composés n'ait pas encore été
identifiée, quelques-uns uns ont été caractérisés dans des études portant notamment sur l'eau
potable. Tousces produits forméspar chloration des eaux sontregroupés sous la dénomination
générale de«sous-produits de chloration »(Chlorinated by-productsouCBPs). Lafigure 1 résumeles réactionscompétitivesdans l'eau douceetdansl'eaudemer
Lessous-produitsde chlorationpeuventêtre divisésen4 classes.
2.1 Leshalogèneslibres
Les espèces les plus représentatives résultant de la dissolution du chlore dans les eaux douces naturelles sont l'acide hypochloreux HCIO et l'anion hypochlorite CIO". Ces deux composés constituentlechlore libre.
Dansl'eaudemer,l'acidehypochloreuxoul'hypochlorite oxydent les bromures dont lateneurest
l'eau de mer (pH = 8), les principales formes réactives en présence sont l'acide hypobromeux (majoritaire)etl'ion
hypobromite.
AminosHalog&nécs NH 2CI NHCI 2 NCI NH 2Br NHBr , Bromamines
\ /
Acidehypochlorcux CIOHt
CIO -+H + Hypochlorite Acidehypobromeux s^BrOH ' BrO -+H+ Hypobromite .ftéfcùrséurs :orgiiiiiqùès:/ \
Organohalogénés volatils Organohalogénés nonvolatilsFigure 1: Réactionsdu chloreeneaudouceeteaudemer
2.2 Leshaloamines
Enprésenced'ammonium ou d'amines organiques,les halogènes libres donnent des chloramines oudesbromamines. La vitesse de formation des monobromamines estdix foissupérieure à celle des chloramines(WajonetMorris, 1980). Il peutseformer des dibromamines ou tribromamines selon des réactions réversibles en fonction du pH (ce qui n'estpas le cas pourles chloramines).
Cependant, les bromamines sont moins stables que les chloramines et se décomposent plus
rapidement dansl'eau. Laformation desbromamines dépend durapportBr2/Netdu pH, alorsque
larépartition des chloraminesdépendde lacinétiquedeformation desespèces (Doré, 1989). Les
halogènes libres réagissent avec la matière organique présente dans l'eau pour former des
composés organohalogénés(OX) qui n'ontpasdepropriétés oxydantes. Les substrats organiques
etles aminessontencompétitionpourles halogènes. AbarnouetMiossec (1992) ontmontréque les rendements deproductionenOXdiminuentlorsque laconcentrationen
NH4+
augmente.2.3 Lestrihalométhanes(THM)
Ce sont les composés organiques volatils générés par la chloration des eaux naturelles. La
formation des THM parchloration deseauxde boissonafait l'objetd'études approfondiesàcause
notamment du risque sanitaire potentiel qu'ils représentent. Dans l'eau de mer chlorée, le bromoformeetle dibromochlorométhanesontlesTHMmajoritaires, le bromoforme constituant à lui seulplus de 90% des THM produits (HelzetHsu, 1978)et(Abarnou, 1979).
2.4 Les AutresOX
Parmi les autres OX identifiés, trois catégories sont fréquemment trouvées dans les eaux naturelles chlorées : les acideshalocicétiques, les haloacétonitrilesetles halophénols.
Nous avons cherché dans cette étude à connaître lacinétique et le rendement de formation des
THM, des haloacétonitriles et halophénols dans des eaux de mer provenant de divers sites de
centrales nucléaires et chlorées expérimentalement au laboratoire ainsi qu'à déterminer les
principaux sous-produits dechloration formés lorsdeschlorationsin situ.
3. Matériel et Méthodes
Cette étudeaété réalisée surdesprélèvements d'eau de mer,effectuésdans les canaux d'amenée
etdes déversoirs de rejet des centrales de Gravelines, Penlyet Paluel situées en Mer du Nordet enManche.
L'eau est prélevée en surface par immersion d'une cage en acier inox contenant des flacons en
verre afin d'éviter les contaminations dues à l'utilisation de matièresplastiques. Toute la verrerie utiliséeestpréalablement nettoyée à l'aide d'un détergent, rincéeabondamment à l'eau du robinet
puis à l'eau déminéralisée. Elleestensuiteséchéedans uneétuve à100°C.
Dans le cadre des essais effectués au laboratoire, la chloration estpratiquéesur les échantillons non filtrés. Le chlore actifest ajoutéaux concentrations désirées parl'addition d'hypochlorite de
sodiumensolution dans de l'eaudéminéralisée, autitre vérifiépariodométrieen milieu acétique. Tous les essais de chlorationontété effectués à 20°C-1.
Les espèces oxydantes ont été neutralisées par ajout d'un excès de thiosulfate de sodium au
moment de l'échantillonnage après des temps de contact variables afin de déterminer les
cinétiques de formation.
Les échantillons pourlamesurede THM sont conservés auréfrigérateur à 4°C dans des flacons
de type pénicilline, munis de septum et sertis après prélèvement. Les THM sont identifiés et
mesurés parchromatographic en phase gazeuse avec un chromatographe HP série 6890 équipé
d'un détecteur à capture d'électrons, spécifique des halogènes, après une extraction de type «
purgeandtrap»(méthode EPA 524-2).
Leshaloacétonitrilessontidentifiésetmesurés parchromatographicenphasegazeuse (H.P. série
6890équipé d'undétecteur à spectrométrie de masse) aprèsuneextractionparde l'acétate d'éthyle
(HodgesonetCohen, 1990).
Les halophénols sont identifiés et mesurés par chromatographic en phase gazeuse (H.P. série
6890 équipé d'un détecteur à spectrométrie de masse) après une extraction par du
dichlorométhane suivie d'une dérivation par l'acide acétique anhydre et d'une concentration
(Soniassyetai, 1994).
Toutescesdifférentes méthodes demesuresontrésumées dans lafigure 2. Le tableau 1 rassemble
les performances analytiques obtenues pour chacune de ces trois familles de composés
THM Haloacétonitriles Halophénols Domaine de linéarité étudié 0-50 ng/1 0-30ng/1 0-10|ig/l Dérives absence absence absence
Etalonnageinterne oui oui oui
Répétabilité 8% 8% 8%
Seuil dedétection 0,05mg/1 0,1 Mg/1 0,05 pg/1
Seuildequantification 0,1pg/1 0,5 |ig/l 0,1 pg/1
Tableau 1 :Performances des méthodes de dosages
Famille THM Principe Haloacétonitriles P&T MicroextractionL/L CPG/ECD CPG/MS Halophénols Extraction L/L Concentration Dérivation CPG/MS Mise en œuvre 5 mleau 48 mleau milieu acide 4 ml acétated'éthyle 500 mleau
dérivationanhydride acétique
extractionCH2C12(90ml)
concentration(1 ml)
Figure2 :Méthodes d'analyses des principaux sous-produits dechlorationutilisées
4.
Résultats
et discussion4.1 Etudeexpérimentale
Cinétiqueetrendement de formation du bromoforme
Laprésence de THM dans les eaux afait l'objet de nombreuses études depuis la publication de
Rook (1974), démontrant leur formation au cours de la chloration des eaux de surface. Elle
s'explique par la réaction haloforme, spécifique des méthylcétones, qui conduit toujours aux
trihalométhanes. Les précurseurs de cette réaction sont évidement les méthylcétones ettous les
composés facilement oxydables en composés possédant un groupement méthylcétone (Doré et
Goichon, 1978). Ces composéssont nombreux (alcools, aldéhydes, P dicétones, polyphénols) et
naturellement abondants. Dans l'eaude mer, où le chloreoxyde instantanément les bromuresen acidehypobromeux (SugametHelz, 1977), (Wonget Davidson, 1977)et(Helzet Hsu, 1978), le bromoforme apparaît comme le produit final d'une suite de réactions comprenant l'oxydation
rapideet totale des bromuresparle chlore, l'oxydation des composés organiques parle chlore et
carbonyl suivie du clivage en milieu basique de la molécule formée. Helz et Hsu (1978) et
Abarnou (1979)ont montréque, lorsde la chlorationdel'eau demer, si lasalinité dépasse 8
%>o,
le bromoformereprésente plus de 90% du total des THMformés.Dans une première série d'essais, nous avons observé l'apparition de trihalométhanes et
l'augmentation de leur concentration lors d'additions croissantes de 0,5 mg/1 à4 mg/1 de chlore
(tableau2).
Les concentrationsen THM sont corrélées avec les quantités de chlore ajouté. Le bromoforme
représente96% des THM formés, le dibromochlorométhane 3%, le dichlorobromométhaneet le
chloroformemoins de 1%. Chlore(mg/1) 0,5 0,7 1 1,2 1,5 1,7 2 2,5 3 4 Chloroforme(p g/1) 0,10 0,07 0,08 0,09 0,10 0,12 0,12 0,15 0,17 0,21 Br-CL- 0,04 0,04 0,05 0,06 0,06 0,09 0,17 0,18 0,30 0,40 méthane(|ig/l) Br2-Cl-méthane 0,28 0,38 0,53 0,61 0,68 0,88 1,34 1,98 4,16 4,70 (Mg/1) Bromoforme(pg/1) 9,32 13,95 21,21 17,07 20,20 24,55 42,27 58,66 91,43 86,91
Tableau2 :Formationdes THMenfonction du chlore ajouté (après24h de contact) dans l'eau demerde Gravelines 28/08/96
(NH/=0,21mg/l)
La formation du bromoforme est un processus rapide aux faibles concentrations en chlore
(< 1mg/1) : laquantité de bromoforme produiteen 3 heuresest pratiquementéquivalente à celle
produiteen 24 heures : 95% pour0,8 mg/1, 82% pour 1,0 mg/1 et65% pour 2,0 mg/1(figure 3).
Eneau douce, lacinétique de formationdu chloroformeestbeaucoup plus lente (Doré M., 1989).
*5. a. E U <2 o S o u. 69 5 10 15 20
Duréede laréaction(heure)
chlorationà0,8ppm;
chloration àlppm chloration à2ppm
Figure3:Cinétiquede formation du bromoformepourdifférentesconcentrations
Le rendement de formation massique du bromoforme (produit majoritaire des THM) est très
faible, de l'ordre de 2%,comme l'indiquelafigure4,avecde légères différences suivant laqualité
de l'eau prélevée. Cette valeur est du même ordre de grandeur que celle trouvée par Abarnou
(1979)déterminée après chloration d'eau demerprélevéeen zonesd'estuaires après 30 minutes. Il serait intéressant de déterminer le rendement de formation du bromoforme dans deseaux de mer
de qualité ou d'origines différentes afin de mieux pouvoir modéliser les quantités de THM
produitsparla chlorationdeseaux de refroidissement.
100,00-r 90,00■■ 80,00 ■-70,00• ■ 60,00■ ■ 50,00■ -40,00■ 30,00 -20,00■ 10,00■ —I 1 I 1 1 1 1 1,00 1,50 2,00 2,50 3,00 3,50 4,00 Chlore(mg/L) +Gravelines28/08/96! «Penly 03/10 96 j APaluel24/04/97
Figuïe4: Concentrationde bromoforme produitetrendement massique
selon letauxde chlore(après24 h). Essaissur eaudemerprélevée à Gravelinesle28/08/96, àPenly le 03/10/96, àPaluel le 24/04/97.
Cinétiqueetrendement dudibromoacétonitrile
La formation du dibromoacétonitrile (DBAN) est un processus lent : la quantité produite en 24 heuresestpratiquement équivalenteà celle produiteen48heures (voirfigure 5).Le rendement de
formationmassiqueestfaible: environ 0,3%.
Cinétiqueetrendement du2,4,6-tribromophénol
Comme pourle dibromoacétonitrile, la cinétique de formation du2,4,6-tribromophénol(TBP)est relativementlente : laquantité de TBP produiteen 8 heures représente 80% de celle produiteen 24 heures pour une chloration à 4 mg/1 et, pour une chloration plus importante à 10 mg/1, elle
représente moins de 70% (voirfigure 6).
Lerendement de formationmassiqueesttrès faible (de l'ordre 0,03%) (voir figure 7).
La faiblesse des rendements montre que la formation des trois familles de sous-produits de
chloration étudiées ne représente qu'unepart infime de la consommation en oxydants. Un travail
importantest donc à réaliser afin de détermineret quantifiertous les autres composés halogénés
z < ca Q 20r 18 16 --14 --12 10 8 6
"t*
4 *. ♦Paluel 10mg/L 08/12/97 AGravelines 4mg/L03/03/98 10 20 30 Tempsenh 40 H 50Figure5: Cinétiquedeformation du DBANpourdifférentesconcentrationsen chloreinjecté. 3,5 D> 3. 3 2,5 c XQ> r. % 2 E o £ 1,5 4tauxde chloration 4mg/L ; ■tauxde chloration 10mg/L\ 1 0,5 0 10 15 Tempsenh 20 25
Figure 7; Concentrationde2,4,6-tribromophénol produitetrendementmassique
selon letauxde chlore(après24h). Essaissur eaudemerprélevée
à Gravelinesle 04/03/98etàPenly le03/03/98
4.2 Suivi de la formation dessous-produits de chloration in situ
Des mesures de THM, haloacétonitriles et halophénols ont été ensuite effectuées sur des
échantillons collectésauniveau de déversoirs derejets des sites de Penly, GravelinesetPaluel de 1995 à 1998.
Seuls le bromoforme, le dibromoacétonitrile et le tribromophénol ont pu être trouvés dans ces
eaux de rejets à des concentrations significativement supérieures aux limites de détections des
méthodes demesure utilisées(tableau3). Cesvaleurssont du même ordre de grandeurquecelles
rapportées parJenneret al. (1996) sur des centrales situées en bordure de la Mer du Nord (les
concentrations en bromoforme pourles sitesde Heysham et Maasvlaktesont respectivement de 25 /jg/1 et 11 pg/1 ; celles du dibromoacétonitrile et du tribromophénol sontde 2,4 jjg/1 et 0,26
|jg/l à Heysham). Chloreinjecté (mg/1) c CHBr3 (Mg/1) o DBAN (m g/i) o TBP (Mg/1) o Gravelines 0,81 0,24 24,72 13,23 3,34 0,89 0,33 0,10 Penly 0,34 0,14 7,17 4,88 0,42 0,08 0,10 -Paluel 0,77 0,03 18,74 5,02 - - -
-Tableau3:concentration moyennedechlore, bromoforme, dibromoacétonitrile
et2,4,6-tribromophénolmesuréssursitesetécarts-type estimés
De plus, les rendements massiques de formation pour ces trois composés sont très proches de ceux déterminés au laboratoire. La répartition moyenne des sous-produits de chloration à
Gravelinesestreprésentée dans la figure 8. Onconstatequele bromoforme constitue le principal
sous-produitde chloration,commecelaaété observé précédemmentlors des essais de chloration
aulaboratoire.
°TBPa autresTHM 10% |0/r
5%
13 Bromoforme
84%
Figure8:Répartitionmoyennedes sous-produits de chloration à Gravelines
(chloration:0,8mgA)
5. Conclusion
La chloration de l'eau demerdonne lieu à la formation dedifférents composéshalogénés dontles
trihalométhanes, les haloacétonitrileset les halophénols que nous avons identifiés et quantifiés
parchromatographicenphase gazeuse. Danschaque famille, on notela forte prédominance d'un
seul composé : bromoforme, dibromoacétonitrile et 2,4,6-tribromophénol. Les cinétiques de formation de ces composés sont relativement rapideset les rendements deformation massiques
sont faibles (<5%). Les rendements massiques de formation mesurés au laboratoire et sursites
sont presque identiques : la chloration en laboratoire de l'eau de mer des canaux d'amenée de
Gravelines, PenlyetPaluel à 1 mg/1 dechlore produitunequantité debromoformecomparable à
celle trouvée lors de campagnes de prélèvements réalisés de 1995 à 1998 sur les rejets de ces centrales nucléaires, chlorés entre 0,3 mg/1 et 1 mg/1, soit entre 7 fag/1 et 30 pg/1. Pour le
dibromoacétonitrile et le 2,4,6-tribromophénol, les rendements massiques de formation sont
également très proches de ceux évalués au laboratoire. Le bromoforme , principal THM formé lors de la chloration de l'eau de mer, représente à lui seul plus de 90% de l'ensemble des
composés dosés par CPG. Les autres sous-produits de chloration (halophénols et
haloacétonitriles) sont présents à des concentrations très faibles dans les eaux de rejet de
centrales, le dibromoacétonitrile et le tribromophénol représentant respectivement à peine 5%et
1% desorganohalogénésidentifiésetquantifiés.
Environ 250tonnes de bromoformesontproduitesparan parla chloration continue des centrales
nucléaires d'EDF situéessurla MancheetMer duNord, ce quinereprésenteraitpasplus de
104
fois laproduction globale annuelle estiméeparles alguesmarines.
Desdonnées écotoxicologiquesvontêtrecollectées afin de mieux prendreen comptel'impact de cessous-produits de chlorationsurlemilieu marin.
Références
bibliographiques
Abarnou A., 1979. Trihalométhanes formés par chloration de l'eau de mer. 2e'""journées de la thermoécologie, Institut scientifiqueettechnique des pêches maritimes, 14-15 novembre 1979
Abarnou A., Miossec L., 1992. Chlorinated waters discharged to the marine environment
chemistryand environmental impact. An overview. The Science of the Total Environment, 126
: 173-197
Doré M., Goichon J., 1978. Etude d'une méthode d'évaluation globale des précurseurs de la
réactionhaloforme. WaterRes., 14,p 657-663
DoréM., 1989. Chimie desoxydantsettraitementdeseaux,Lavoisier,p 143-326
HelzG., Hsu R., 1978. Volatile chloro- and bromocarbons in coastal waters.Limnol. Oceanogr.,
23, 5 : 858-869
Hodgeson J., Cohen A., 1990. Determination of chlorination desinfection by products and
chlorinated solvents in drinkingwaterby LL extraction and GC with ECD. Method EPA 551 : 169-200
JennerH.,Taylor C., Van Donk M., Khalanski M., 1996. Chlorination by-products in chlorinated coolingwaterofsome europeancoastalpowerstations. Marine Environmental Research, 43, 4
: 279-293
Morizur Y., 1985. Moyens de lutte contre les salissures marines. Rapport IFREMER
85/DIT-GO/R05 : 1-25
Rook J. J., 1974 . Formation of haloforms during chloration of natural water. Wat Treat. Exam.,
23 : 2 34-243
Soniassy R., Sandra P., Schlett C., 1994. Water Analysis, ed Hewlett-Packart Compagny, Germany, 278p
Sugam R., Helz G., 1977. Speciation of chlorine produced oxydant in marinewaters : theoretical
aspects. ChesapeakeScience, 18, 1 : 116-118
Wajon J., Morris J., 1980. Bromaminationchemistry : rates of formation of NH2Br and some
N-bromamino acids, in Water Chlorination-EnvironmentalImpactand Health Effects, volume 3; Ann Arbor Science : 171-181
WongG., Davidson J., 1977. The fate of chlorine inseawater. WaterResearch, 11 : 971-978
OXYDATION DE MOLECULES
ORGANIQUES
DISSOUTES AU COURS DES TRAITEMENTS DE L'EAU :
ROLE DES REACTIONS RADICALAIRES
AnneVENTURA,Valérie CAMEL, Alain BERMOND
Institut NationalAgronomiqueParis-Grignon, Laboratoire de Chimie Analytique, 16rueClaude Bernard75005Paris(France)
Résumé
La potabilisation d'une eau de surface impose l'élimination d'une partie importante de la matièreorganiqueprésente. L'oxydationparl'ozone constitue l'une des étapes du traitement
quipermet d'atteindre cetobjectif. Toutefois, certaines molécules ne sontpas oxydées dans les conditions classiques d'ozonation. Elles nécessitentpar conséquent une oxydation plus
poussée, réalisée grâce à la génération dans l'eau à traiter de radicaux hydroxyles OH°, dont
le pouvoir oxydant est très élevé. Plusieurs procédés assurent cette génération ; les plus répandus sont le couplage ozone-peroxyde d'hydrogène et le réactif ferreux-peroxyde d'hydrogène (plusconnu sous le nom réactifde Fenton). Pour cedernier, la production de
radicauxOH° résulte de l'oxydation du fer ferreux en fer ferrique par le peroxyde
d'hydrogène. Ces oxydants combinés présentent malgré tout un certain nombre de
désavantages. En particulier, leur coûtde mise en œuvre estrelativement élevéen raison de
l'utilisation du peroxyde d'hydrogène. De plus, dans le cas du réactif de Fenton, on peut
observer un épuisement progressif de la concentration enfer ferreux, celle-ci devenant le
facteur limitant de la réaction. C'est pourquoi les méthodes électrochimiques ont déjà été
utilisées afin de générer continûment le fer ferreux à partir du fer ferrique et le peroxyde
d'hydrogène à partir de l'oxygène dissous. La génération électrochimique du réactif de
Fenton (électro-Fenton) présente des avantages majeurs : (1) aucun ajout de réactif mis à
partunequantitécatalytique defer, d'où uncoût réduit; (2)parles faibles concentrations de
ferferrique misesen œuvre, unesolubilité du fer jusqu'à pH 5;(3)lapossibilitéthéorique de quantifier le nombre de OH° formés grâceausuivide l'intensitéau coursde la réaction (si le
fer ferriqueuniquementestréduit).
Les résultatsprésentéssontobtenusavec un colorant, l'azocarmine B, choisicomme modèle
simple de la matière organique naturelle. Enpremier lieu, l'oxydation a été conduite à un
potentiel (E = 215 mVAg/AgCi) où seul le fer ferrique est réduit, en présence de peroxyde
d'hydrogène. Diverses concentrations de feret de peroxyde d'hydrogène ont été testées, et
lesperformancessontcomparées àcelles du réactif de Fenton. Ensuite, les résultats obtenus lors de la génération simultanée du fer ferreux et duperoxyde d'hydrogène sontprésentés
(E--200BiVAg/Agciy Ces essais, ainsi que quelques tests préliminaires réalisés sur
l'atrazine, montrent les ressources importantes de la génération électrochimique de
radicaux.
Motsclés:Oxydation,Fenton, électro-Fenton, radicaux
1. Contexte
La matière organique présente dans les eaux brutes destinées à être potabilisées doit être éliminée en grande partie lors du traitement de potabilisation, afin de distribuer une eau
conformeà laréglementation etde limiter la reviviscencebactérienne dans le réseau. Pour les eaux uséesonestégalement confrontéà la nécessité dedégraderla matièreorganique afin de
limiter son rejet dans le milieu naturel. Cette élimination peut être assurée par une étape
d'oxydationchimique. L'ozoneestgénéralementretenucommeoxydanten raison desonfort
pouvoiroxydant. Malgrétout, certaines molécules sontréfractaires à l'oxydation parl'ozone
seule (c'est parexemple le cas de l'atrazine) ; il est donc préférable de mettre en œuvredes
procédés d'oxydation avancée, c'est-à-dire dessystèmesgénérant l'entité radicalaire OH° très
oxydante. Les systèmes les plus utilisés à l'heure actuelle sont le couplage ozone-peroxyde
d'hydrogène, et le réactif de Fenton (Fe(II)-H202). Toutefois ces systèmes présentent un
certain nombred'inconvénients,enparticulieruncoûtélevéenraison de l'ajout de H2O2,etla
difficulté de maîtriser le nombre de radicaux formés (et donc par voie de conséquence les
réactions se déroulant dans le milieu). En outre, le réactif de Fenton souffre d'un
appauvrissement en fer ferreux (en raison de son oxydation en fer ferrique, et de sa lente
régénérationenfer ferreux ensuite) (Al-HayeketDoré, 1985) ;enfin,sonefficacité imposeun
pH voisin de 3. C'est pourquoi l'utilisation d'un système électrochimique assurant une
génération in situ du fer ferreux à partir de fer ferrique est particulièrement séduisante. En
outre, il est également possible de générer le peroxyde d'hydrogène dans le milieu par réduction de l'oxygène dissous. Nous avons appliqué ce système (désigné sous le nom
d'électro-Fenton) à l'oxydation de deux molécules modèles : l'azocarmine B (un colorant,
modèle simple de la matière organique naturelle) et l'atrazine (herbicide fréquemment rencontré dans leseaux desurface, dont le schéma dedégradation par les radicauxestconnu, l'acidecyanurique constituant le sous-produit d'oxydationultime). Les résultats obtenus ont
été, àchaque fois, comparés àceux obtenusavecleréactif de Fenton classique.
2. Le
système Fer-H202
2.1 Lagénération électrochimique du réactifdeFenton
Plusieurs systèmes électrochimiques ont été utilisés pour générer le réactif de Fenton. Les
principaux sont rassemblés dans le Tableau I; le plus souvent ils mettent en œuvre des cathodesen mercure.
Le ferferreuxestproduitparréduction du ferferrique :
Fe3++e" Fe2+
Leperoxyde d'hydrogèneestsoit rajouté directement danslemilieu,soitgénéréparréduction del'oxygène dissous, selon la réaction:
Application Naturede la Source deFe2+ Source de H202 Référence
cathode
Hydroxylation de l'acide Nappe de Hg Réduction de Fe+ Réduction de 02 OturanetPinson
benzoïque ouPt (1995)
Hydroxylation del'acide Nappede Hg Réduction de
Fe3+
Ajout de H2O2ou Oturanetal. (1992)salicylique ouajout deFe2+ réduction de 02
Hydroxylation du benzène Nappe de Hg Pas de ferou Ajout de H2O2ou Fleszaretal. (1977)
etduphénol réduction deFe3+ réduction de 02
Hydroxylation du benzène Nappede Hg,Pb Pasde fer,ouajout Réductionde 02 FleszaretSobkowiak
etduphénol ouCu deFe2* (1983)
Dégradation de l'anilineet Carbone-PTFE* Pasde fer Réduction de 02 Brillasetal. (1995)
de la alimentéeen02
4-chloroaniline
Oxydation d'eaux usées de Plaqueenacier Anodeenfer Réduction de O2 LinetPeng (1996)
l'industrie textile inoxydable
Oxydation d'eaux usées de Fer Anodeenfer Ajoutde H202 LinetChen(1997)
l'industrie textile
*
PTFE:polytétrafluoroéthylène.
Tableau I:Principalesapplicationsdelagénération électrochimiquedu réactif
deFenton.
Une application a montréquel'hydroxylation du benzèneestpossible lors de la réduction de
l'oxygène en l'absence de fer dans le milieu (Fleszar et Sobkowiak, 1983). L'ajout de fer
permet évidemment d'accélérer la réaction, en raison d'une génération plus facile des radicaux hydroxyles. D'autres résultats sur la dégradation de l'anilineet de la chloroaniline confirment la formation de radicaux hydroxyles en l'absence de fer(Brillas etal., 1995). La réaction d'initiation des radicauxproposéeserait la suivante :
H20 -» OH°+H++e"
Les radicaux formés peuvent ensuite réagir avec le peroxyde d'hydrogène pour former de nouveauxradicaux:
H202+0H°-> HO2°+H2O
Ladégradation des composésorganiquespeutalorssefaire àpartir de l'actiondecesradicaux
(Brillasetal., 1995) :
OH°+RH -> H20+R°
H02O+RH -> H2O2+R°
Dans l'étude que nous avons entreprise, nous avonschoisi demettreen œuvrela réduction du
ferferrique à une nappede mercure,leperoxyde d'hydrogèneétant soit rajouté à la solution, soit produitsimultanémentparréductiondel'oxygènedissous.
2.2 Principalesréactions radicalaires
Les réactions intervenant dans le système Fenton classique sont bien connues (Walling,
1975), (Walling et Cleary, 1977). La réaction initiale est l'oxydation du fer ferreux par le peroxyded'hydrogène:
(1)
Fe2+
+H202 ->Fe3+
+0H°+OH" k,=76M"'.s"'
Elle est suivie d'un certain nombre de réactions (Walling, 1975), (Sun et Pignatello, 1993),
(ChenetPignatello, 1997):
(2) OH°+Fe2+ -> Fe3+ +OH" k2=4.3
108 M"1.s"1
(3) Fe2++H02° ->
Fe3+
+H02" k3= 1.2106
M"1.s"1
(4) Fe3++H02° -4
Fe2+
+02+H+ Lt= 1104M"'.s"'
(en milieu acide)(5) Fe3++H202 -4
Fe2+
+H02° +H+ ks=0.02M"1.s"1
(en milieu acide)(6) OH°+OH° H202 ke=5.3
109 M"'.s"1
(7) 0H°+H202 -4 H20+H02° k7=2.7
107 M"1.s"1
(8) HO2°+HO2° -4 H202+02 kg=8.5
105
M"1.s"1
En présence du substrat organique (noté R ou RH), on a les réactions (Al-Hayek et Doré,
1985): (9) OH°+R—> ROH° (10) OH°+RH —» H2O+R° (11) R°+O2h> R02o (12) R02°+H20-4 R0H+H02° (13) R02°+
Fe2+
R02"+Fe3+
(14) R°+Fe3+
—> R++Fe2+
Lorsde lagénération électrochimiquedu fer ferreuxà partir deferferrique,on a enplus :
(15)
Fe3+
+e"Fe2+
En présence deperoxyde d'hydrogène,on a donc la réaction de Fenton (réaction (1)) quise
produit. Lorsqu'onréduit simultanément l'oxygène, onréalise la réaction suivante (Oturanet Pinson, 1995):
(16) 02+2H++2e" H202
Figure 1:Réactions intervenantenprésencedu systèmeélectro-Fenton.
3. Matériel et Méthodes
3.1 Réactifs utilisés
Les solutions des composés étudiés sont réalisées à partir des produits sous forme solide
(azocarmine B, C28HnN3Na209S3: Fluka ; atrazine et simazine: certifiées à 97%, CIL Cluzeau).
Le fer ferreux et le fer ferrique sont ajoutés sous forme de sulfate de fer, respectivement
(FeS04,7 H2O) (Merck, qualité pour analyse) et Fe2(S04)3 (Normapur, Prolabo). Le pH des
solutions estajusté à 3 avec del'acide sulfurique(H2S04, RP Normapurpour analyses, 95%,
Prolabo).
Le mercureutilisé estdequalitépourla polarographie (Merck). Le peroxyde d'hydrogèneest sousforme de solution stabilisée à 30% (Merck).
3.2 Celluleélectrochimique
La cellule (25 mL)comprenduneconnexion métalliqueen sonfond. Une agitationencontinu assurel'homogénéisation de la solution.
Les troisélectrodes utiliséessontlessuivantes: • électrode de travail :
nappedemercure(surface voisinede 12
cm2)
• électrode auxiliaire : fjldeplatine • électrode de référence:
Les électrodes auxiliaires et de réféience sont placées dans un compartiment, rempli de
solution aqueuse (pH 3).
Lorsdes réactionsd'électro-Fenton, unpotentielestimposéentre l'électrode deréférenceet
l'électrode de travail, grâce àpotentiostat (Tacussel ASA 100-1). Lecourantentre l'électrode
auxiliaire et l'électrode de travail estsuivi ; dans lecas où seul le fer ferrique estréduit, ce
courantestproportionnel à la concentrationenfer ferrique dans la solution.
Lorsdes réactions deFenton, on mesurelepotentielentre l'électrodede travailetl'électrode
de référence ; l'application de la loi de Nernst permet de connaître les concentrations
respectives de ferferreuxetferrique.
3.3 Dégradation de l'azacarmineB
La dégradation de l'azocarmine B a été suivie en continu, grâce au montage expérimental
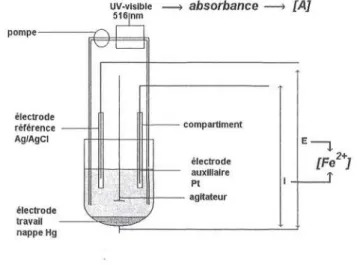
présentésurlafigure2. Unepompe(Beckman, modèle 110A)assurela circulationencontinu
de la solution à travers la cellule d'un spectrophotomètre UV-visible (Jasco 875-UV). La mesurede l'absorbance à516nra permet,grâce àunétalonnage, de connaître la concentration d'azocarmine Bnondégradéeàun instant donné.
pompe-électrode référence Ag/AgCI UV-visible 516|nm absorbance [A] -compartiment électrode auxiliaire Pt agitateur rr- 2+t [Fg ]
Figure 2 :Montage expérimental utilisépourlesuivide l'oxydation de
l'azocarmine B.
3.4 Dégradation de l'atrazine
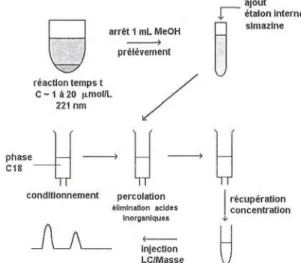
Contrairement à l'azocarmine B, l'atrazine ne peut être suivie en continu par simple
spectrophotométrie car les produits d'oxydation de cette molécule présentent un maximum
d'absorbance à la même longueur d'onde (221 nm). Par conséquent, il est nécessaire de réaliser au préalable une séparation chromatographique. Ceci a été réalisé par
chromatographic en phase liquide (LC) (pompe microgradient Applied Biosystems 1410;
vanne d'injection Rheodyne 8125, 20 |iL) couplée à un spectrophotomètre (Applied
Biosystems785A ; 221 nm)etàunspectromètre demasse(MS) (PE SciexAPI 100)viaune interface électrospray. La séparation est effectuée sur une colonne de silice greffée C|«
(Spherisorb, 100mm x 1.0 mmi.d., 5 pm), avec un mélangeméthanol/eau (acidifiéspar0.04
% d'acide tormique)50/50 (70
|iL.rnin"').
L'utilisation du spectromètre de masse impose l'élimination des sels inorganiques de la solution. Pourcela,une extractionen phase solidesurcartouches Cis (Supelclean C18, 1 g, 6
mL) aétéréalisée,selon lesconditionssuivantes : conditionnementavec2 mL de méthanol, 2 mL d'acétate d'éthyle, 2 mL de méthanol, et 2 mL d'eau (pH 3) ; passage de l'échantillon aqueux (15 mL) ; séchage pendant 15 min à l'air ; élution avec2,5 mL de méthanol. Après
ajout de 2,5 mL d'eau, l'extrait est injecté en LC-MS. Nous avons opéré par étalonnage
interne pour quantifier l'atrazine, en choisissant la simazine comme étalon ; celui-ci a été
rajouté à la solution avant l'extraction en phase solide, afin de s'affranchir des possibles
perteslors du traitement de l'échantillon (voir figure 3).
Aprèschaqueprélèvement àuntemps tdonné, la réactionaété stoppéeparl'ajout de 1 mL de
méthanol(cesolvant étantunpiège à radicaux).
ajout étalon interne ^ , t* simazine arret 1 mLMeOH Mg;/ prélèvement réactiontemps t C«1à 20 n.mol/L 221nm phase C18 " conditionnement
J\JV
TT percolation élimination acides Inorganiques Injection LC/Masse TT récupération concentration \JFigure 3: Traitementde l'échantillonavantanalysede l'atrazineparLC-MS.
4. Résultats et discussion
4.1 Dégradation de l'azoearmine
Réduction du ferferriqueenprésencede peroxyded'hydrogène
La réduction du ferferrique seul est réalisée à un potentiel de + 215 mV. Les suivis de la
disparition de l'azoearmine B pour les trois systèmes, Fenton, électro-Fenton et le témoin
(Fe(III)-H202sanspotentiel) pour unrapport[H202]/[Fe]=lsontsimilaires (voir figure 4) ;on observe àchaquefois unpallier, pourenviron 50% d'abattement de l'azoearmine B. Le suivi du fer ferreuxau coursdecesréactionsestdonnéfigure5.
[azo B] (mg/L) 15 EF 10 — 5 0 temps(min) 50 100
Figure4:Dégradation de l'azocarmineB
enprésenced'unrapport[H2O2] /[Fe] =1.
EF: électro-Fenton(à+215 mV);F:Fenton; T: témoin
Conditionsexpérimentales:pH=3 ;[H2O2] =[Fe] = [azo B]=
3.10'5
M.Figure 5:Suivi dufer ferreux lors de la dégradation de l'azocarmineB
enprésence d'unrapport[H2O2] /[Fe]= 1.
EF: électro-Fenton(à +215 mV); F:Fenton;T: témoin
Conditionsexpérimentales:pH=3; [H2O2]=[Fe] -[azo B] =
3.10'5
M.Au cours de la réaction de Fenton on observe l'appauvrissement en fer ferreux, et
parallèlement laformation de fer ferreux au coursdela réactiond'électro-Fenton. Initialement
le fer ferreux est généré dans le milieu; au début il réagit avec H2O2 et redonne du fer
ferrique; par la suite, la teneur en H2O2 diminue, et du fer ferreux s'accumule dans la
solution. On remarque que pour le témoin
(Fe3+-H202,
en l'absence de réactionélectrochimique) le fer ferreux est également généré dans le milieu, ce qui conduit à un
abattement comparable àceux par Fenton et électro-Fenton. Par ailleurs, on peut noter un excès de fer ferreux pour la réaction de Fenton, ce qui suggère que l'abattement limité de
l'azocarmine B résulte d'une limitation en H2O2. Pour confirmercette hypothèse, les mêmes
expériences ont été réalisées avec un excès de peroxyde d'hydrogène ([H2C>2]/[Fe]=10). Les résultats sontdonnésfigure 6.
[azo B] (mg/L)
Figure6:Dégradation de l'azocarmine B
enprésence d'unrapport[H2O2] /[Fe] =10
EF: électro-Fenton (à +215 mV); F:Fenton; T: témoin
Conditionsexpérimentales:pH=3 ;[H2O2] =
3.10'4
M;[Fe] =[azo B] =3.10'5
MDanscesconditions l'azocarmine Besttotalementdégradéeparleréactifde Fenton. Enoutre, si le réactif de Fenton est initialement le plus rapide (en raison d'un nombre de radicaux initialement générés très important), les performances des deux systèmes Fenton et électro-Fenton sont sensiblementcomparables parla suite. A nouveau la,dégradation du colorant en
présence du témoinesttrès importante. Ilestvraisemblableque pourle témoin ilseproduiten
premierlaréaction (5), suivie de la réaction (4),cequi génère du ferferreux dans le milieu ;
celui-ci peut ensuite réagir avec H2O2 pour redonner des radicaux. De plus, la structure moléculaire complexe de l'azocarmine B laisse à penser que des produits d'oxydation de celle-ci favorisent la génération du fer ferreux dans le milieu, selon la réaction (14) ; en effet
certains composés aromatiques (quinones) favorisent cette réaction (Chen et Pignatello.
1997). D'autres études ont déjàmontré l'abattement possible de molécules organiques par le
système
Fe3+-H2Û2
à pH 3 (Pignatello et Huang, 1993), (Sun et Pignatello, 1993), (Chen etPignatello, 1997).Le suivi dufer ferreux dans la solutionestdonné figure 7.
Figure7:Suividu ferferreux lorsde la dégradation de l'azocarmineB
enprésenced'un rapportavec[H2O2]/[Fe] - 10.
EF: électro-Fenton(à +215 mV);F:Fenton; T: témoin
Cette figure montre l'appauvrissement important en fer ferreux au cours de la réaction de Fenton (qui a lieu plus rapidement que précédemment), ainsi que l'accumulation de fer
ferreuxavecles systèmes électro-Fentonet
Fe3+-H202.
Réduction du ferferriqueet réductionsimultanée de l'oxygène
Laréduction simultanée du fer ferriqueet de l'oxygèneest réalisée à-200 mV. La figure 8
montie ladiminution de la concentration en azocarmine B pour les trois systèmes suivants :
électro-fenton, H2O2,etH2O2 (à-200 mV).
Figure8:Dégradation de l'azocarmine Blors de la génération électrochimique
deFe2+etH2O2.
EF: électro-Fenton(à -200 mV); T1 :H2O2 ; T2:H2O2 (à-200 mV)
Conditionsexpérimentales:pH= 3; [H2O2]=
3.10"1
M;[Fe3+]
= [azo B]=3.10'5
M.Onconstatetoutd'abord l'efficacitéimportante du système électro-Fenton,cequi confirmela
potentialité de ce système pour le traitement de l'eau. On observe une cinétique de dégradation comparable à celles obtenues précédemment en présence de peroxyde d'hydrogène ou avec le réactif de Fenton. Parcontre on note une dégradation relativement importante de l'azocarmine B aveclesystème électrochimique en l'absence de fer (H2O2 à -200 mV). Dansce cas, onpeutenvisager l'une des deux réactions de génération des
radicaux!-hydroxyles :
H20-^ OH°+H1"+e"
H2O2+e"—> OH°+OH"
Ainsil'oxydation de l'eau (à une électrode de platine) aété utilisée afin d'oxyder le phénol
(Comninellis etPulgarin, 1991). Demême, l'hydroxylation du phénol enprésence de H1O1 (à
unpotentiel réducteur)etsans ajout de fer semble être liée à la décomposition catalytique de
H2O2 à la surface de l'électrode (Fleszar et ai.. 1977). Par ailleurs, il n'estpas exclu que la présence detracesde fer ait contribué àune génération radicalaire dans la solution.
4.2 Dégradation del'atrazine
Laréduction du fer ferriqueen présence de peroxyde d'hydrogène aété testée surl'atrazine,
etcomparéeauréactifclassique de Fenton, ainsi qu'autémoin
(Fe3+-H202).
Commelemontrelafigure9, le système électro-Fenton offre à nouveau des performances comparables à celles du réactif classique. En revanche, on constate une faible dégradation par le système
Fe"+-H2C>2, ce qui confirme le rôle joué par les produits de dégradation de l'azocarmine B
dans lagénérationdes radicaux.
temps(min)
Figure 9:Dégradationde l'atrazineenprésenced'unrapport[H2O2] /[Fe] = 10.
EF: électro-Fenton(à+ 215 mV); F:Fenton; T: témoin
Conditionsexpérimentales:pH- 3; [H2O2] =3.10'
[Fe] =[atrazine]
M,
3.ia5M.
5. Conclusion
Cette étudepréliminairemontrelafaisabilitéde larégénération électrochimiquedu fer ferreux
pour dégrader par oxydation de type Fenton des molécules organiques. L'efficacité de ce
procédé dépendcependant de nombreux facteurs; enparticulierellepeutêtre limitée lorsque
lesproduits d'oxydation participent à la régénération du fer ferreux (cas de l'azocarmine B). De nombreuses perspectives d'application de ce procédé au traitement des eaux sont envisageables. Pour des raisons de coût, elles font appel à la réduction simultanée du fer
ferrique enfer ferreuxet de l'oxygène dissousen peroxyde d'hydrogène. Par ailleurs, il sera
REFERENCES
BIBLIOGRAPHIQUES
AL-HAYEK N., DORE M., 1985. Oxidation des composés organiques par le réactif de
Fenton: possibilitésetlimites. Environ. Technol. Lett., 6, 37-50.
BRILLAS E„ BASTIDA R. M.,LLOSA E„ CASADO J., 1995. Electrochemical destruction
of aniline and 4-chloroaniline for wastewater treatment using a carbon-PTFE 02-fed
cathode. J. Electrochem.Soc., 142, 1733-1741.
CHEN R., PIGNATELLO J. J., 1997. Role of quinone intermediates as electron shuttles in Fenton and photoassisted Fenton oxidations of aromatic compounds. Environ. Sci.
Technol., 31, 2399-2406.
COMNINELLIS C., PULGARIN C., 1991. Anodic oxidation of phenol for waste water
treatment.J.Appl. Electrochem., 21, 703-708.
FLESZAR B., SOBKOWIAK A., 1983. Hydroxylation of benzene and phenol during
electroreduction of oxygen.Electrochim.Acta,28, 1315-1318.
FLESZAR B„ SOBKOWIAK A., KOWALSKI J„ 1977. Electrochemical investigations on reactions of benzene andphenol with hydroxyl radicals.Roczn. Chem., 51, 339-344. LIN S. H., PENG C. F., 1996. Continuous treatment of textile wastewater by combined
coagulation, electrochemical oxidation and activated sludge. Wat. Res., 30, 587-592.
LIN S. H., CHEN M. L., 1997. Textile wastewater treatment by enhanced electrochemical
method and ionexchange. Environ. Technol., 18, 739-746.
OTURAN M. A., PINSON J„ BIZOT J., DEPREZ D„ TERLAIN B„ 1992. Reaction of
inflammation inhibitors with chemically and electrochemically generated hydroxyl radicals. J. Electroanal. Chem., 334, 103-109.
OTURAN M. A., PINSON J. , 1995. Hydroxylation of electrochemically generated OH°
radicals. Mono- and polyhydroxylation of benzoic acid : products and isomer's distribution. J. Phys. Chem., 99, 13948-13954.
PIGNATELLO J.J., HUANG L. Q., 1993. Degradation of polychlorinated dibenzo-p-dioxin
and dibenzofuran contaminants in 2,4,5-T by photoassisted iron-catalyzed hydrogen
peroxide. Wat. Res., 27, 1731-1736.
SUN Y., PIGNATELLO J. J., 1993. Photochemical reactions involved in the total
mineralization of2,4-Dby
Fe3+/H202/UV.
Environ. Sci. Technol., 27, 304-310.WALLING C. , 1975. Fenton's reagentrevisited.Acc. Chem. Res.,8. 125-131,
WALLINGC., CLEARY M. , 1977. Oxygen evolution as acritical testof mechanism in the
EVOLUTION DE LA MATIERE
ORGANIQUE
DEGRADATION DE LA MATIERE
ORGANIQUE
ET
PROCESSUS
BIOGEOCHIMIQUES
MIS EN JEU
DANS UNE BERGE DE L'ESSONNE
CyrilMoneron, Jean-MarieMouchel, CEREVE (ENPC-ENGREF-UPVM),
6et8avenueBiaisePascal, CitéDescartes, ChampssurMarne,
77455 Marne la Vallée Cedex2.
Tél:01 64 1536 16, Fax: 01 64 15 37 64, Email:moneron@cereve.enpc.fr
Résumé
Les processusde transformation desnutrimentsauseind'une bergeontété étudiéssur unsite
représentatifdesfonds de vallées crayeuses à tourbe, nombreuses dans le bassin parisien.
Uneparticularité de ce site est un sens d'écoulement de l'eau orienté de la rivière vers la
berge; ainsi, la qualité de l'eau estparfaitement connue à son entrée dans la berge. Des cellules à dialyses ont été insérées horizontalement dans la berge pour comprendre les
processus de transformation de l'azoteet du phosphoreetles interactions qui les relient à la
circulationde l'eauainsiqu'auxcycles de l'oxygène, du carbone, du feretdu manganèse. L'eaude la rivières'infiltre horizontalementdans la berge à des vitessesvariantde 9,3
cm.j'1
en hiver à 66
cm.j'1
en été. Les profils de concentration obtenus dans l'eau interstitielletraduisent une variabilité temporelle importante. Alors que les gradients de concentrations
sont beaucoup plus marqués au mois de novembre (83 mg-N-NÛ3
.U'.m1)
qu'au mois dejuillet (37 mg-N-NÛ3
.L~'.m'
), les flux d'élimination denitratessontenjuillet supérieurs d'un facteur 8,2à ceuxde novembre, respectivement de 2,46et 0,3 g-N-NO3.m2.jour1.
L'activitémoyennede la biomasseest4,4 fois plus élevée enjuillet qu'en novembre, passantde24,6à
6,0g-N-NO3
.f'.m3.
Parconséquent, l'efficacité dénitrifiante dece milieu estessentiellementliée à la vitesse d'écoulement de l'eau, et à un moindre niveau à l'activité saisonnière de la
biomasse..
Une augmentation inattendue des teneurs en carbone organique dissous dans l'eau interstitielle estobservée enjuillet. Tenantcomptedesprocessus consommateursde carbone
(respiration aérobie, dénitrification, réduction des hydroxydes de fer et manganèse), la production totale de carboneorganiqueestestimée à30
g.m3.f'.
En regard des réserves decarboneorganique disponibles dans la berge (12
kg-COT.m3),
cette quantité représenteraitune consommation mensuelle de 13,5 % du stock total de carbone de la berge. Une telle
consommation ne peut s'expliquer que par une accumulation de carbone non ou peu
dégradable en hiver ou par une surproduction estivale de carbone organique dissous assimilable, d'origine végétaleoubactérienne.
La comparaison des flux d'ammonium etd'orthophosphates révèle l'importance quantitative de processus affectant les quantités d'orthophosphates autres que la minéralisation de la
matière organique. L'évolution comparée des concentrations en
PO/'
et enFe2+
permetdemettre en évidence une réduction du matériel minéral et notamment des hydroxydes de fer