Savant Genome Browser 2: visualization and analysis for population-scale genomics
7
0
0
Texte intégral
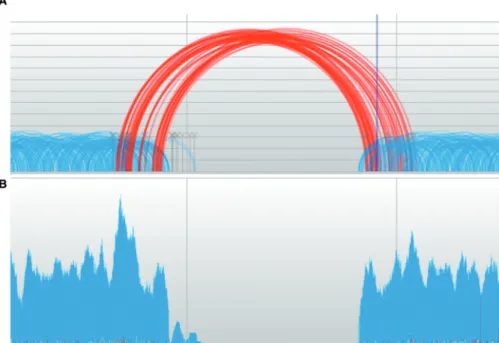
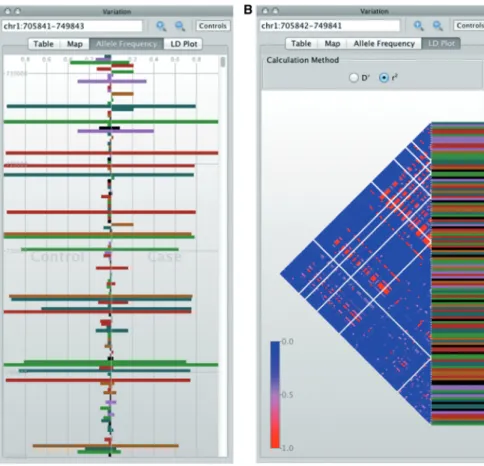
Figure

Documents relatifs
