Combinatorial drug discovery in nanoliter droplets
Texte intégral
Figure
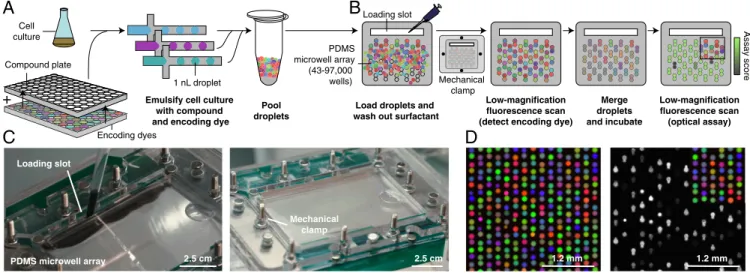
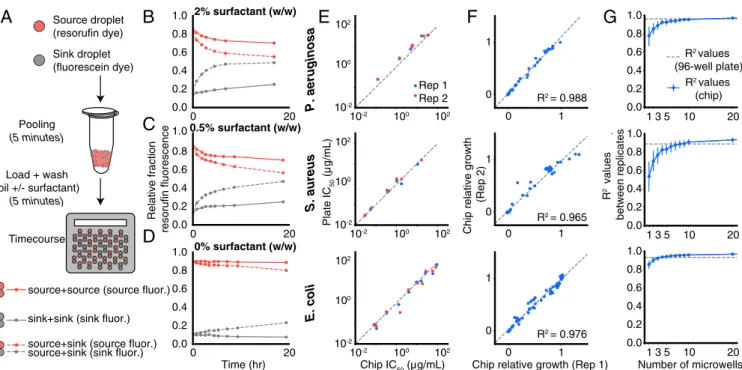

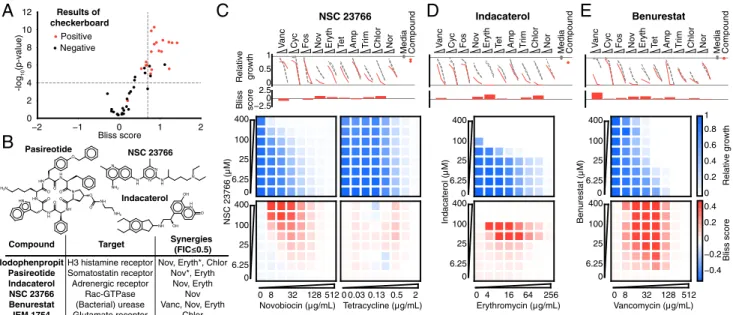
Documents relatifs
For any starting point in R d , d ≥ 3, we identify the stochastic dierential equa- tion of distorted Brownian motion with respect to a certain discontinuous Muck- enhoupt A 2
A Bayesian dose-finding design for drug combination clinical trials based on the logistic model.. Marie-Karelle Riviere, Ying Yuan, Frédéric Dubois,
Wages, as stated in our paper, we used the POCRM design starting at the lowest dose level, based on current practice in Phase I oncology clinical trial.. The authors
Combination of High Dose Hypofractionated Radiotherapy with Anti- PD1 Single Dose Immunotherapy Leads to a Th1 Immune Activation Resulting in a Complete Clinical Response in a
These results are in perfect agreement with the Landauer principle: in a quasi-static process, the mean work or heat required to erase 1 bit of information is ln 2 = 0.693.
Measure each segment in centimeters.. ©c i2Q0y1N1P nK8urtPal kS3oIfLtRwEa0rmeH 6LxLMCm.o m fAAlQla WrqiNgEhxtts4 drUeOsEeir6vje1dV.J U 0MzavdEeD ewDiRtehI
SR 8 -IS-A-HYPERNYM RULE: Any noun compound Arg 1 +Arg 2 made up of Arg 1 whose T(Arg 1 )=Lexical Hierarchy contains the initial part of T(Arg 2 )=Lexical Hierarchy of Arg 2
Appendix 3 (continued): Overview of initiatives supporting antibiotic R&D InitiativeDescriptionAB R&D incentivesValue chain targetedTimeframeAB R&Dfunding EU
