Le syndrome des antiphospholipides (SAPL) est aujourd’hui reconnu comme l’une des plus fréquentes causes de thrombophilie acquise, survenant le plus souvent dans le cadre d’un LES ou isolément (forme primaire). Ce syndrome associe des manifestations cliniques à type d’événements thrombotiques récurrents (veineux ou artériels) ou des complications obstétricales variées, à la présence durable d’anticorps antiphospholipides (aPL).
Les aPL sont un groupe hétérogène d’autoanticorps, traditionnellement subdivisés en lupus anticoagulant et anticorps anticardiolipine en fonction de la méthode de détection, respectivement les tests de coagulation dépendants des phospholipides et la technique immunoenzymatique utilisant la cardiolipine immobilisée. Les véritables cibles des aPL potentiellement thrombogènes sont constituées, non par les phospholipides eux-mêmes, mais par des protéines plasmatiques qui leur sont associées, particulièrement 2–Glycoprotéine I et la
prothrombine. L’amélioration du diagnostic et des approches thérapeutiques du SAPL passe par une meilleure compréhension de l’origine et de la pathogénicité de ces anticorps.
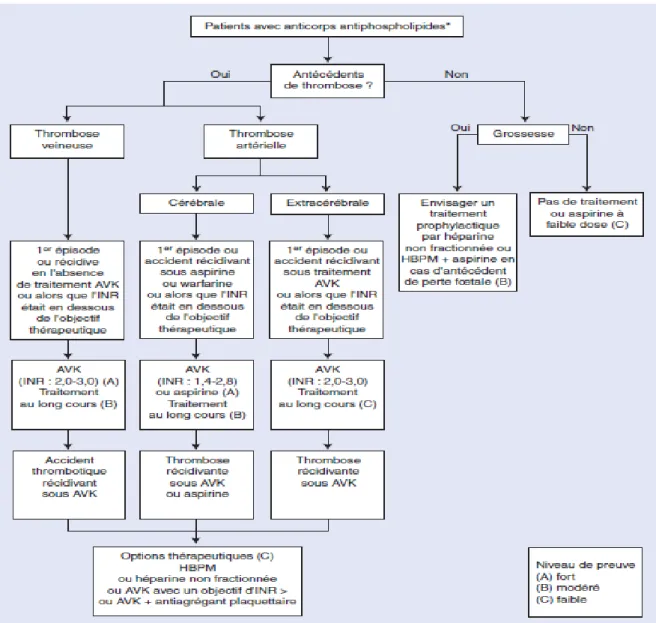
En raison d’un risque majeur de récidive thrombotique dans les années qui suivent l’épisode initial, la prévention secondaire des thromboses veineuses et artérielles par une anticoagulation prolongée est nécessaire. L’objectif thérapeutique est d’avoir un INR compris entre 2 et 3 et au-delà en cas de récidive sous traitement par AVK. Les autres facteurs de risque cardiovasculaires associés (particulièrement hypertension, hypercholestérolémie, tabac) doivent être corrigés en évitant chez la femme d’utiliser les contraceptifs oraux contenant des œstrogènes. L’association d’aspirine à faible dose et
d’héparine permet de prévenir les complications obstétricales associées aux aPL, notamment les avortements spontanés.
L’objectif de notre travail est double :
Rapporter les aspects diagnostics récents du SAPL
PARTIE I
Aspects moléculaires,
épidémiologiques et
I. HISTORIQUE
Il a été rapporté, il y a plus de 50 ans, chez des patients atteints de lupus érythémateux systémique (LES), des cas de positivité dissociée de la sérologie syphilitique, liée au fait que le réactif VDRL contient de la cardiolipine [1].
En 1963, ont été décrits des cas de thromboses associées à un allongement des tests de coagulation lié à la présence d’un anticorps circulant de type lupique, ou lupus anticoagulant (LA) [2].
L’association de fausses couches répétées et d’événements thrombotiques en présence d’un LA a été rapportée dès 1980 par Soulier et Boffa [3]
.
En 1983, Harris et al [4] retrouvent dans le LES une association thrombose et présence d’anticorps anticardiolipine (aCL) détectés par ELISA.
Entre 1983 et 1986, de nombreuses publications, font état de différentes manifestations cliniques paraissant rattachées à ces différents variétés d’anticorps antiphospholipides (aPL) : infarctus cérébral, thrombose des artères rénales ou hépatiques, hypertension artérielle pulmonaire, thrombopénie, livedo, myélite transverse, syndrome de Guillain- Barré [5, 6]. Alors que la première conférence internationale sur les aPL a été organisée à Londres en 1984.
Le SAPL est finalement défini par Harris et al en 1987 comme l’association d’au moins une manifestation clinique à une anomalie biologique. Le SAPL s’individualise du LES dans les années 1987-1988 pour rapidement gagner son autonomie.
Les années 1990 sont marquées par la découverte de « cofacteurs », protéines associées aux phospholipides qui constitueraient en fait la véritable cible des anticorps [7]. Parmi les différents cofacteurs identifiés, la 2
glycoprotéine I ( 2–GpI) est la principale cible des aCL et de certains LA. La
présence chez certains malades ayant fait des thromboses récidivantes d’anticorps anti- 2-GpI isolés a fait proposer le terme de syndrome des
antiphospholipides/cofacteur [8]. Cette situation apparaît néanmoins particulièrement rare.
La fréquence des aCL dans la population générale et l’augmentation de leur titre avec l’âge [9]
peuvent amener à des diagnostics par excès. C’est pourquoi Alarcon-Segovia [10] a proposé des niveaux différents de probabilité diagnostique : SAPL défini, probable et douteux selon le nombre de manifestations cliniques et les taux d’aPL. De nombreuses situations cliniques peuvent s’accompagner d’aPL (Tableau I), mais en dehors du SAPL primaire et des aPL associés au lupus, ces anticorps sont rarement symptomatiques. Des aPL isolés, totalement asymptomatiques, peuvent être découverts à l’occasion d’un bilan de coagulation préopératoire, d’une consultation prénuptiale ou dans le cadre de l’enquête familiale d’un patient ayant un SAPL défini [11]
. Il n’y a pas lieu de parler de SAPL en l’absence d’événement thrombotique et/ou obstétrical. Les critères révisés en 1999 dits de Sapporo ont fait l’objet au congrès de Sydney d’une actualisation récente [12]
Tableau I : Situations cliniques associées aux antiphospholipides (aPL) [11] SAPL primaire
SAPL associé au LES Présence isolée d’aPL
Sujets asymptomatiques, notamment entourage familial de SAPL Maladies auto-immunes
LES sans SAPL
Polyarthrite rhizomélique, maladie de Horton/pseudopolyarthrite
rhizomélique, sclérodermie, syndrome de Gougerot-Sjögren, polychondrite, thrombopénie auto-immune, thyroïdite, sclérose en plaques
Traitement inducteur
Procaïnamide, phénothiazines, hydantoïnes, quinidine, hydralazine, -bloquants, interféron a...
Infections
Viroses aiguës, VIH, hépatite C, syphilis, maladie de Lyme, tuberculose, paludisme...
Cancers solides, hémopathies malignes, immunoglobulines monoclonales Divers
Sarcoïdose, maladie de Crohn, spondylarthropathies, diabète insulinodépendant, insuffisance rénale terminale, insuffisance hépatocellulaire aiguë, éthylisme chronique, maladie périodique, stérilité, CIVD
II. DEFINITIONS
II.1. Anticorps antiphospholipides [13]
Le terme «anticorps antiphospholipides» (aPL) au sens strict regroupe une large famille d’anticorps reconnaissant aussi bien des phospholipides anioniques que neutres (Tableau II). Ces phospholipides sont pour la plupart des constituants des membranes plasmiques cellulaires et sont organisés en bicouche avec une distribution asymétrique entre la membrane externe et interne (Fig. 1).
Le nom des aPL a été élargi à des anticorps dont les cibles antigéniques sont non seulement les phospholipides eux-mêmes («vrais» aPL), mais aussi des protéines plasmatiques associées à ces phospholipides ou ces protéines seules (Fig. 1).
Parmi les aPL, ceux dits «conventionnels», Lupus anticoagulants (LA) et anticorps anticardiolipine (anticardiolipide en français) (aCL), sont classiquement associés au syndrome des antiphospholipides (SAPL) et, bien que non spécifiques du SAPL, ils constituent les critères biologiques pour le diagnostic de ce syndrome.
Tableau II : Les phospholipides cibles des aPL [13].
Figure 1 : Diversité des cibles antigéniques des aPL [13]
Anioniques Neutres Cardiolipine Phosphatidylsérine Acide phosphatidique Phosphatidylinositol Phosphatidylglycérol Phosphatidyléthanolamine Sphingomyéline Phosphatidylcholine
◗ Les LA
Le terme de LA désigne des anticorps définis par leur capacité à prolonger certains tests de coagulation dépendants des phospholipides. Il est admis que ces anticorps reconnaissent des cofacteurs protéiques liés aux phospholipides anioniques. Les principaux cofacteurs des LA sont la bêta2-glycoprotéine I ( 2-GpI) et la prothrombine. Il est important de noter que les LA et les aPL mis
en évidence par des techniques immunoenzymatiques peuvent être des entités distinctes. Bien que ces anticorps soient fréquemment associés au cours du SAPL, leur taux de recouvrement n'est que de 60 % et donc, il conviendra de rechercher la présence d’aPL avec des tests immunologiques et des tests d’hémostase.
◗ Les aCL
La cardiolipine est un phospholipide anionique: constituant de la membrane interne des mitochondries, et récemment, sa présence a été établie dans le plasma sous forme complexée à des lipoprotéines [14] et à la surface de cellules apoptotiques [15]. Les aCL reconnaissent la cardiolipine mais aussi les autres phospholipides anioniques : phosphatidylglycérol, phosphatidylinositol et phosphatidylsérine. C’est la forme oxydée de la cardiolipine qui serait principalement reconnue par les aCL réagissant avec des néo-épitopes présents sur la cardiolipine oxydée [16]. Il convient de distinguer les aCL pour lesquels la réactivité vis-à-vis de la cardiolipine n’est pas dépendante de la présence d’un cofacteur plasmatique dans le milieu réactionnel («vrais» aCL) de ceux «dépendants» parce qu’ils reconnaissent un complexe cardiolipine-cofacteur,
voire même le cofacteur lui-même. Les premiers sont essentiellement retrouvés au cours d’infections alors que les autres sont présents au cours de maladies autoimmunes, dont le SAPL. La 2–GpI, protéine plasmatique appelée aussi
apolipoprotéine H, a été identifiée comme le principal cofacteur des aCL [17].
II.2. Syndrome des anticorps antiphospholipides (SAPL)
Le SAPL est une thrombophilie acquise de définition clinico-biologique, caractérisée par un risque augmentée de thromboses et de complications obstétricales. Cette définition, initialement empirique, basée sur des observations cliniques s’appuie actuellement sur des critères de classification régulièrement révisés par un consensus d’experts [18]
.
En effet, les critères révisés en 1999 dits de Sapporo ont fait l’objet en 2006 d’une actualisation lors du consensus international de Sydney [18]
. Ainsi selon ces nouveaux critères la présence d’un SAPL peut être affirmée devant l’association d’une au moins des manifestations cliniques caractéristiques et la mise en évidence sur le plan biologique d’aPL par une technique de référence (Tableau III) [19].
Les manifestations cliniques retenues dans les critères de classifications sont : les thromboses (artérielles, veineuses, microciculatoires) et les manifestations obstétricales (une ou plusieurs morts fœtales inexpliquées à partir de la 10ème semaine de gestation, une ou plusieurs naissances prématurées avant la 34ème en lien avec une insuffisance vasculaire placentaire, ou au moins 3 avortements spontanés consécutifs avant la 10ème semaine de gestation) [19].
Tableau III : Critères de classification révisés du syndrome des aPL
(critères de Sydney) [19].
Critères cliniques
Thromboses vasculaires
Un ou plusieurs épisodes symptomatiques de thrombose artérielle, veineuse ou d’un petit vaisseau dans n’importe quel tissu ou organe. Cette thrombose doit être objectivée par une stratégie diagnostique validée (c’est-à-dire confirmée par un aspect caractéristique d’un examen d’imagerie de référence ou d’un examen histopathologique). Dans cette dernière situation, il doit s’agir d’une thrombose sans inflammation significative de la paroi vasculaire.
Manifestations obstétricales
(a) Une ou plusieurs morts inexpliquées, fœtus morphologiquement normal, à la 10ème semaine de gestation ou au-delà (morphologie normale établie par échographie ou examen direct) ou
(b) Une ou plusieurs naissances prématurées d’un nouveau-né morphologiquement normal avant la 34ème semaine de gestation à cause :
d’une éclampsie ou d’une prééclampsie grave, ou de signes reconnus d’insuffisance placentaire.
ou
(c) Au moins 3 avortements spontanés consécutifs avant la 10ème semaine de gestation sans cause anatomique ou hormonale maternelle et sans cause chromosomique maternelle ou paternelle
Critères biologiques
(Confirmés au moins 2 fois à une distance de 6 semaines ou plus) 1. Présence d’un anticoagulant circulant (« lupus anticoagulant »-LA), à 2 reprises au moins, espacées de 12 semaines, mis en évidence en suivant les recommandations de l’International Society on Thrombosis and Haemostasis (ISTH).
2. Anticorps anticardiolines (aCL) d’isotype IgG et/ ou IgM dans le sérum ou le plasma, avec un titre moyen ou élevé (> 40 GPL ou MPL, ou >99e percentile), présents à 2 reprises au moins avec des mesures espacées de 12 semaines ou plus, par ELISA standardisé.
3. Anticorps anti- 2 GPI d’isotype IgG et/ ou IgM dans le sérum ou le plasma, avec un titre moyen ou élevé (> 40 GPL ou MPL, ou >99e percentile), présents à 2 reprises au moins avec des mesures espacées de 12 semaines ou plus, par ELISA standardisé.
Le SAPL initialement décrit au cours du LES peut aussi survenir en dehors de tout contexte pathologique auto-immun. Ainsi on distingue :
D’une part, un syndrome primaire (SAPL I) caractérisé par l’association des anomalies cliniques et biologiques présentées dans le tableau III, mais sans aucune maladie auto-immune associée. C’est la forme la plus fréquente du SAPL (53%) [20].
D’autre part, un syndrome secondaire (SAPL II) associé à une maladie auto-immune (47%), essentiellement à un lupus systémique (37%) (Tableau IV). Il faut bien distinguer la rareté du syndrome clinico-biologique de la fréquence du phénomène biologique (aPL) qui n’est pas une anomalie spécifique [20].
Tableau IV : Critères d’exclusion du SAPL primaire [20].
La présence de l’un de ces critères n’est pas compatible avec le diagnostic de SAPL primaire :
Eruption malaire. Lupus discoïde.
Ulcération orale ou pharyngée (sauf ulcération ou perforation de la cloison nasale).
Arthrite franche.
Pleurésie en l’absence d’embolie pulmonaire ou d’insuffisance cardiaque gauche.
Péricardite en, l’absence d’infarctus du myocarde ou d’insuffisance rénale marquée.
Protéinurie supérieure à 0.5g/ jour due à une glomérulonéphrite par complexes immuns prouvée histologiquement.
Lymphopénie < 1000/µl
Anticorps anti-ADN natif, par radio-immunologie ou immunofluorescence sur Crithidia
Anticorps anti-antigènes nucléaires solubles. Anticorps anti-nucléaires à un titre > 1/320. Traitement connu comme inducteur d’aPL.
III. PHYSIOPATHOLOGIE
Le SAPL appartient à la catégorie des maladies auto-immunes ou les manifestations cliniques sont directement attribuables au caractère pathogène des autoanticorps. Cependant l’hétérogénéité clinique du SAPL suggérer l’existence chez les patients d’autoanticorps responsables par des mécanismes distincts, des effets cliniques observés [21].
III.1. Cofacteurs protéiques des anticorps antiphospholipides
Il est généralement admis que les antiphospholipides sont faussement nommés puisque dans la majorité des cas, ils reconnaissent non pas les phospholipides seuls mais des complexes composés de protéines sériques (appelés cofacteurs) et de phospholipides anioniques sont essentiellement des cardiolipines, les phosphatidylsérines et des phosphatidyléthanolamines. En pratique, les cofacteurs protéiques ont la propriété commune de fixer les phospholipides anioniques, créant ainsi des complexes reconnus par les autoanticorps appelés antiphospholipides. Ainsi l’anticoagulant circulant de type lupique, qui reste encore le meilleur témoin du risque thrombotique, correspondre à des autoanticorps antiphospholipides reconnaissant le complexe prothrombinase constitué des facteurs Xa, V et prothrombine liés à des phospholipides anioniques. Ces cofacteurs comprennent entre autres la 2–GpI,
la prothrombine, la protéine C, la protéine S, la thrombomoduline et l’annexineV. Toutes ces protéines sont impliquées dans les mécanismes physiologiques de contrôle de la coagulation et on comprend mieux que la présence d’auto anticorps dirigés contre des complexes associant
pretéines-phospholipides anioniques est susceptible d’interférer avec les mécanismes naturels anticoagulants et de promouvoir la thrombose [21].
III.1.1. Rôle de la β2-GPI et des anticorps anti-β2GPI
La β2-GPI est un cofacteur plasmatique capable d’interagir avec la phosphatidylsérine membranaire pour former un complexe protéine cofacteur/phospholipide. Composée de cinq domaines repliés en sushi, cette protéine de 326 acides aminés possède sur le cinquième domaine une séquence cationique capable de fixer les phospholipides. Les principaux épitopes reconnus par les anticorps correspondants sont plutôt disposés sur les domaines I et IV, mais la discussion n’est pas définitivement tranchée [22]
.
Cette protéine pouvait être le cofacteur principal au cours du SAPL, mais il existe toujours un doute sur le rôle pathogène direct des autoanticorps
anti-2GpI. Cependant, dès à présent, la recherche de ces autoanticorps est incluse
dans les nouveaux critères de diagnostic du SAPL [21].
Ces auto-anticorps anti- 2–GpI illustrent assez bien les difficultés rencontrées
dans tentatives faites pour établir des corrélations entre un événement clinique et la présence d’autoanticorps. En effet, les anti- 2–GpI s’avèrent eux-mêmes
hétérogènes puisque seuls qui reconnaissent le premier domaine protéique de la
2–GpI auraient une activité de type anticoagulant circulant et pouvaient
provoquer des thromboses. Les tests classiques de détection des anti- 2GpI ne
permettent pas de repérer spécifiquement les autoanticorps reconnaissant ce premier domaine protéique [21]. En effet, les rares déficits congénitaux en β2-GPI
ne sont pas associés à une thrombophilie. Cependant, on lui attribue de nombreuses propriétés résumées dans le Tableau V.
L’existence d’un ou plusieurs récepteurs de surface pour la β2-GPI n’est pas totalement éclaircie et plusieurs protéines ou protéoglycanes semblent pouvoir remplir cette fonction de cofacteur d’adhésion : citons l’héparane sulfate, la mégaline (composant endocytique de la famille des récepteurs LDL) et l’APO ER2, enfin, l’annexine II, récepteur endothélial du tissue plasminogen activator (t-PA). Les aPL sont susceptibles d’activer les cellules endothéliales comme l’atteste l’augmentation de production de NF-k B et de la MAPK p38, mais le blocage de ces deux voies ne suffit pas à abolir l’effet des aPL (en particulier les anti-β2GPI) [23]. L’activation par les IgG anti- β2GPI implique la mise en jeu de la protéine adaptatrice MyD88 et TRAF6 et une phosphorylation d’IL1-receptor activated kinase (IRAK) suggérant un mode d’activation analogue à celui induit par le lipopolyosaccharide bactérien (LPS) via le récepteur TLR4 ou l’IL1 via l’IL1-R [24]
.
En résumé, l’hypothèse prévalente actuelle serait que la β2-GPI fixée à l’héparane sulfate à la surface des cellules endothéliales serait reconnue par TLR-4 du fait d’une structure commune entre β2-GPI et certains motifs bactériens ou viraux [25, 26]. Il en résulterait une activation de la cellule endothéliale. Une interaction avec l’annexine II pourrait également déclencher cette fixation sur TLR-4 [27].
III.1.2. Prothrombine et anti-prothrombine (PT)
Il existe plusieurs catégories d’anticorps antiprothrombine : certains reconnaissent le complexe phospholipide/PT, d’autres la PT seule [28]
. La fréquence de tels anticorps est élevée en présence de LA, mais leur valeur prédictive d’un risque de thrombose est très inconstante, très inférieure à celle des anti-β2GPI [27].
Tableau V : Principales fonctions de la β2GPI [27]
Inhibition de l’activation par contact de la voie intrinsèque de coagulation Inhibition de l’activité prothrombinase des plaquettes
Inhibition de l’agrégation plaquettaire à l’ADP
Inhibition de l’activation du facteur XII par kaolin ou acide éllagique Inhibition de l’activation de la prékallicréine médiée par le FXIIa Inhibition de la génération du FXa par les plaquettes
Inhibition de l’autoactivation du FXII plasmatique
Inhibition du complexe prothrombinase FVa dépendant et potentialisation de la production de thrombine en présence de protéine C activée
Inhibition de l’activation procoagulante en inhibant l’activité de la protéine C activée
Inhibition de la protéine S libre (active) en favorisant sa liaison à la C4BP Favorise l’épuration des cellules en apoptose.
III.2. Rôle de l’annexine V et de l’annexine II et anticorps correspondants
Plusieurs milliers de protéines différentes partageant un domaine répétitif (habituellement répété quatre fois), d’environ 70 acides aminés, constituent la famille des « annexines ». La partie aminoacide N-terminale porte les propriétés spécifiques à chacune de ces protéines [27].
III.2.1. Annexine V
L’annexine V a une forme de disque concave avec un domaine de fixation pour les phospholipides membranaires et un autre pour la fixation du calcium. Cette protéine est capable de former un film continu cristallin à la surface des cellules, formant une barrière avec les fluides extracellulaires, empêchant, entre autre, l’interaction avec les protéines de la coagulation/ fibrinolyse. L’annexine V est ainsi synthétisée en abondance par les cellules endothéliales vasculaires et les cellules du trophoblaste. En l’absence de ce film continu, l’annexine V et les facteurs de coagulation vont pouvoir s’activer en cascade, notamment après expression membranaire du facteur tissulaire (TF) sur les phospholipides de membrane (PL) (Fig. 2) [29]. Le complexe TF/PL fixe le facteur VII qui s’active (VIIa) sur le complexe qui se fixe sur le facteur X et le facteur IX pour générer les enzymes Xa et IXa. Chaque enzyme clive son substrat, respectivement le facteur II (prothrombine) et le facteur X en présence de phospholipides anioniques telle la phosphatidylsérine. Ainsi se forme la prothrombinase (complexe formé des facteurs IXa–VIIa–X appelé aussi ténase) qui augmente la production de Xa. La thrombine générée (IIa) transforme le fibrinogène en fibrine, mais ce n’est pas sa seule propriété. L’expression du TF endothélial persiste souvent longtemps après l’épisode thrombotique chez les patients avec
un SAPL. Certaines statines (fluvastatine et simvastatine) ont la propriété, in vitro, d’inhiber l’expression du TF par les cellules endothéliales stimulées par les IgG antiphospholipides et pourraient constituer un traitement adjuvant utile du SAPL [30]. Rand et Wu [31] ont montré que la β2-GPI et les autoanticorps (IgG) correspondant interféraient avec la fixation de l’annexine V à la surface des phospholipides membranaires des cellules trophoblastiques en culture, des cellules endothéliales en culture (cellules de veine de cordon ombilical HUVEC) et des plaquettes. Il en résulte une diminution de la quantité d’annexine V sur ces cellules [32]. Les temps de coagulation in vitro de plasma au contact de cellules trophoblastiques sont raccourcis en présence d’IgG antiphospholipides, cela résultant du déplacement de l’annexine V de la surface cellulaire. Les antiphospholipides agissent soit directement, soit via la formation d’un complexe avec le cofacteur β2-GPI [33]. Plusieurs travaux ont étudié le rôle des anticorps antiannexine V dans les pertes foetales du lupus et les accidents de thrombose : il semble exister une relation avec les pertes fœtales, mais les résultats concernant l’association au risque de thrombose sont divergents et aucune conclusion n’est possible [27]
Figure 2 : Activation de la coagulation par le TF associé aux phospholipides de
III.2.2. Annexine II
L’annexine II est le récepteur membranaire du t-PA, protéine clé dans la fibrinolyse qui, une fois activée, va transformer le plasminogène en plasmine. Il a été montré que l’annexine II fixait aussi la β2-GPI avec une forte affinité [34]
. Des anticorps anti-annexine II ont été mis en évidence avec une fréquence significativement plus élevée dans le SAPL avec thrombose veineuse (17,5 %) ou artérielle (34,3 %) ou mixte (40,4 %), comparativement aux témoins (2,1 %) dans une série de patients mexicains [35]. L’activation de la coagulation induite par les antiannexine II est de même ordre de grandeur que celle obtenue par les anti-β2GPI en conjonction avec la β2-GPI [36].
III.3. Rôle du complément
C’est au travers d’un modèle murin de SAPL obstétrical que G. Girardi et al ont démontré le rôle prépondérant de la cascade du complément à l’origine des lésions placentaires, des pertes fœtales et retard de croissance induit par les aPL. Ainsi, les souris Balb/C, âgées de deux à trois mois gestantes de 8 à 12 jours, qui reçoivent des injections intrapéritonéales d’IgG humaines antiphospholipides (10 mg) vont développer un retard de développement et des résorptions fœtales nombreuses comparées aux souris injectées avec des IgG normales. L’implication du complément est attestée par l’absence de résorption fœtale si les souris sont Knock-out pour le gène du C3 ou si elles reçoivent simultanément du Crry-Ig (3 mg tous les deux jours de j8 à j12). Cette protéine de fusion s’oppose à l’action de la C3 convertase classique et alterne, bloquant ainsi l’activation du complément [37]
même équipe a montré le rôle primordial des deux voies du complément, la voie classique (les souris déficitaires en C4 sont protégées) et la voie alterne d’amplification (les souris déficitaires en facteur B sont aussi protégées). Il est possible d’inhiber le SAPL obstétrical dans ce modèle murin en utilisant des souris génétiquement déficitaires en C5 ou en C5aR (récepteur du C5a présent sur les polynucléaires, les monocytes ou les plaquettes).
Seules les IgG antiphospholipides complètes (avec partie Fab et Fc) sont efficaces dans cette activation du complément, la partie F (ab’) 2 étant sans effet. C’est le site de fixation/activation du complément qui est important et non la capacité à se fixer sur les Fcγ récepteurs [38]
(Fig. 3). Dans un modèle murin de thrombose induite par striction de la veine fémorale et injection d’IgG humaines antiphospholipides, il a été possible de mettre en évidence le même rôle de l’activation du complément. Les souris déficitaires en C4 sont protégées, de même qu’un prétraitement par un anticorps monoclonal anti-C5 [39]
. Parmi les aPL, les IgG anti-β2GPI se sont montrées capables d’induire l’activation du complément dans un modèle de thrombose mésentérique induite par l’injection conjointe d’antiphospholipides chez des rats préalablement sensibilisés par une injection de LPS [40]. En aval de cette activation complémentaire par les antiphospholipides fixés sur les membranes trophoblastiques ou vasculaires, deux phénomènes semblent survenir : une production accrue de TNFα dans le tissu décidual qui se traduit par des taux sériques augmentés [41] et une libération importante de récepteur soluble de type 1 du VEGF (sVEGFR-1) ou sFlt-1, molécule puissamment antiangiogénique [42], ce qui va freiner le développement normal du placenta, et conduire aux complications obstétricales. L’héparine est un puissant anticoagulant et son utilisation a été recommandée sur cette
propriété pour traiter le SAPL thrombotique et obstétrical lorsque ce dernier résiste à l’acide acétylsalicylique à dose antiagrégante. L’héparine est également un puissant agent inhibiteur du complément et l’équipe de G. Girardi et al [43]
a démontré que c’était cette propriété anticomplémentaire qui expliquait son efficacité clinique, qu’il s’agisse des HNF ou des HBPM. En effet, d’autres antithrombotiques puissants, tels le fondaparinux ou les hirudines sont incapables de prévenir les pertes fœtales du SAPL murin. Cette propriété conforte la pratique clinique actuelle et si le modèle est transposable à l’homme, c’est un encouragement à utiliser plus précocement les HBPM dans les SAPL obstétricaux [27].
Figure 3 : Rôle de l’activation du complément dans les pertes fœtales induites
III.4. La diversité des structures moléculaires des aPL
La structure moléculaire des aPL est extrêmement variée, non seulement en terme de classe d’immunoglobulines (IgA, IgM ou IgG), mais de façon plus importante en terme de leurs régions variables d’immunoglobulines qui sont responsables de la liaison à l’autoantigène. Une analyse systématique des régions variables de chaînes lourdes et légères d’anticorps monoclonaux antiphospholipides provenant de différents patients révèle que ces anticorps utilisent une large fraction des gènes V disponibles dans le répertoire de gènes V humains [44,45]. Récemment il a été montré que cette variabilité des aPL est également considérable à l’échelon d’un seul patient : les lymphocytes B produisant des antiphospholipides provenant d’une patiente atteinte de SAPL primaire ont été triés et les régions variables d’immunoglobulines des anticorps produits ont été analysées après amplification génique. Parmi les nombreuses régions variables de chaînes lourdes et de chaînes légères analysées nous n’avons pas trouvé de séquences identiques ou similaires. De plus non seulement ces régions variables étaient très différentes d’un aPL à un autre, mais encore certains de ces aPL (essentiellement de classe IgG) étaient mutés somatiquement, suggérant que les lymphocytes B producteurs avaient été soumis à une pression de sélection positive par l’autoantigène [46]
.
Récemment, chez l’homme, il a été montré que les lymphocytes B producteurs d’aPL sont présents dans le répertoire périphérique du sujet normal, peuvent produire des aPL extrêmement variés, et comprennent un groupe modeste de cellules B mémoire capables de produire des formes mutées somatiquement de ces autoanticorps [47]. Ainsi, chez les adultes sains, les lymphocytes B produisant des aPL de faible affinité peuvent maturer
normalement dans la moelle osseuse, migrer en périphérie où ils peuvent rejoindre les centres germinatifs et devenir des lymphocytes B mémoire producteurs d’aPL. Si l’on s’attache aux hypothèses actuelles, les aPL normaux, en raison de leurs capacités à se lier aux phospholipides anioniques, participent à la clairance des cellules apoptotiques et des débris apoptotiques [48]. Pour être plus précis, l’importance réelle des aPL dans cette fonction est actuellement très mal définie comparée à d’autres mécanismes qui participent à l’élimination des particules et des cellules apoptotiques [49]. Cependant, dans des conditions normales, ces cellules B produisant des aPL sont immunologiquement ignorantes (elles ne sont pas spontanément activables par leur antigène), ne produisent pas de taux significatif d’aPL, mais peuvent devenir transitoirement activées et produire des aPL au cours, par exemple, des pathologies infectieuses.
Les liens éventuels entre ces lymphocytes B producteurs d’aPL physiologiques et les lymphocytes B producteurs d’aPL pathogéniques, ne sont pas clairs mais peuvent être explorés par l’utilisation de techniques de transfert passif d’immunoglobulines antiphospholipides purifiées dans des modèles murins de thrombose ou de grossesse [50]. En effet, différents travaux ont montré que des aPL purifiés provenant de patients atteints de SAPL étaient capables de reproduire des pertes fœtales et des résorptions fœtales chez les souris enceintes. Utilisant ce modèle, J. Salmon a démontré l’importance cruciale de l’activation du complément par sa voie classique au cours de la phase effectrice de la pathologie placentaire induite par les aPL [51].
IV. ÉPIDEMIOLOGIE
La prévalence du syndrome des aPL dans la population générale est faible. Seul un faible pourcentage de patients qui présentent une thrombose veineuse ou artérielle ont des aPL à un titre significatif et persistant. Il ne faut pas confondre la fréquence des aPL décrits dans différentes populations et celle du SAPL.
Dans les populations générales, les études rapportant une fréquence élevée d’antiphospholipides sont souvent biaisées et tiennent compte de faibles concentrations d’anticorps ou d’anticorps non persistants (notamment post-infectieux). En dehors du lupus systémique, la prévalence des anticorps est faible. Cependant, elle peut atteindre 40 % des patients au sein d’une population de lupiques. Dans les 2 types de situations, la fréquence des thromboses est la plus élevée lorsque l’aPL est un anticoagulant circulant. Si l’on prend l’exemple d’un premier épisode de thrombose veineuse, le risque relatif associé à un anticoagulant circulant est de 6 ce qui dépasse nettement le risque associé à un aCL ou à un anticorps anti- 2-GpI. Le risque de récidive thromboembolique
veineux est augmenté chez les patients qui présentent simultanément des anticoagulants circulants et des anticorps anti- 2-GpI [19].
PARTIE II
I. LES THROMBOSES ARTERIELLES ET VEINEUSES [1]
La thrombose observée au cours du SAPL a la particularité de survenir sur une paroi vasculaire saine, indemne de toute infiltration cellulaire. Parfois, le thrombus se constitue sur une lésion athéromateuse, les aPL apparaissent alors comme un facteur précipitant. Tous les territoires vasculaires peuvent être touchés : artères, artérioles, capillaires, veinules, veines profondes ou veines superficielles.
I.1. Thromboses veineuses
Ce sont de loin les plus fréquentes. Les territoires profonds veineux des membres inférieurs sont plus souvent concernés, mais tous les sites sont possibles. L’attention doit être attirée vers un SAPL d’autant plus que la thrombose veineuse survient dans un territoire inhabituel : veine cave supérieure ou inférieure, veines rénales, veines surrénales, veines mésentériques, veine porte ou veines sus-hépatiques, veines rétiniennes ou veines des sinus veineux cérébraux ou veines superficielles, en l’absence de varices. La présence d’aPL constitue non seulement un risque de premier épisode thrombotique veineux, mais aussi un risque important de récidive. Associé au LES, le SAPL peut s’exprimer pour la première fois à tout âge. La thrombose veineuse est toujours multifactorielle, c’est pourquoi il ne faudrait pas, sous prétexte d’une grossesse, d’un alitement ou de la prise d’oestroprogestatifs, négliger la recherche d’aPL si le phénomène thrombotique survient avant l’âge de 50 ans et bien sûr si un LES est associé.
I.2. Thromboses artérielles
La thrombose peut concerner tous les territoires artériels quel que soit le calibre vasculaire, des gros vaisseaux à la microcirculation. Le système nerveux central est plus fréquemment concerné. Il peut s’agir d’accidents ischémiques transitoires ou constitués. Le territoire carotidien est plus souvent touché que le territoire vertébrobasilaire. Le syndrome de Sneddon est une entité particulière du sujet jeune qui associe livedo et infarctus cérébral. Les aPL y sont présents près d’une fois sur deux et le risque de récidive est important en l’absence de traitement avec une évolution possible vers la démence vasculaire .L’imagerie par résonance magnétique nucléaire (IRM) cérébrale avec séquences T2 et séquences Fluid Attenuated Inversion Recovery (FLAIR) est l’examen de choix pour mettre en évidence les infarctus cérébraux (parfois silencieux). L’IRM retrouve aussi assez fréquemment des hypersignaux de petite taille (de signification incertaine) dans la substance blanche corticale ou souscorticale et plus rarement une atrophie cérébrale. Les aPL sont reconnus aujourd’hui comme un véritable facteur de risque indépendant d’accident ischémique cérébral avec un risque important de récidive rapidement après un premier épisode en l’absence de traitement. Ce risque chez un individu donné est huit fois plus élevé lorsque des aPL sont présents. De nombreuses manifestations neurologiques ont été rapportées associées aux aPL. Toutes les manifestations rapportées n’ont cependant pas un support thrombotique, il s’agit parfois de simple association comme par exemple au cours de la fibrose multiloculaire. En cas d’infarctus cérébral, le mécanisme peut être embolique à point de départ cardiaque. L’échodoppler cardiaque fait donc partie des examens nécessaires et indispensables dans le SAPL avec thrombose artérielle.
II. MANIFESTATIONS CARDIAQUES
Les principales manifestations cardiaques rapportées en association aux aPL sont les anomalies valvulaires, les thromboses coronaires et l’athérosclérose coronaire, les hypertrophies ventriculaires et dysfonctions ventriculaires, les thrombi intracardiaques, l’hypertension artérielle pulmonaire et les complications cardiaques du syndrome catastrophique du SAPL.
II.1. Atteintes valvulaires
L’atteinte valvulaire cardiaque est la plus fréquente des manifestations cardiaques. Elle est décrite chez 11,6 % des 1000 patients de l’étude Euro-APS
[52,53], soulignant la fréquence d’une telle atteinte au cours du SAPL primaire et
rapportant 32 à 38 % de lésions valvulaires pour moins de 5 % des témoins. Dans cette même étude, les résultats pour le SAPL secondaire au LES sont comparables et retrouvent 14 à 65 % d’atteintes valvulaires contre 0 à 40 % chez les témoins. En utilisant l’échographie transoesophagienne, technique plus sensible pour dépister les atteintes valvulaires, Turiel et al retrouvaient des anomalies valvulaires chez 31 des 40 patients étudiés atteints de SAPL (82 %).
Plusieurs études ont montré une forte association entre l’existence de lésions valvulaires et la survenue d’infarctus cérébraux [54]
. Les anomalies valvulaires retrouvées sont assez similaires à celles observées au cours du LES : épaississement valvulaire, lésions nodulaires irrégulières, végétations, fuite ou sténose. L’atteinte valvulaire est d’autre part significativement corrélée au titre des aCL. Les atteintes valvulaires sont par fréquence surtout mitrales puis aortiques [55]. L’endocardite « verruqueuse atypique » de Libman-Sachs,
considérée comme caractéristique du LES, est aujourd’hui plutôt assimilée à une atteinte liée aux aPL.
Le suivi évolutif des patients atteints de SAPL est aussi particulièrement intéressant [55]. Dans ce travail, les auteurs ont fait une échocardiographie transoesophagienne à 56 de leurs patients atteints de SAPL, un épaississement valvulaire était retrouvé chez 34 d’entre eux (61 %), cinq avaient un aspect d’endocardite de Libman-Sachs. Au cours des cinq années de suivi, trois patients sont décédés (un de complication hémorragique après valvuloplastie, un d’infarctus du myocarde et un de défaillance viscérale multiple avec coagulation intravasculaire disséminée [CIVD]). À 5 ans, l’échocardiographie était inchangée chez 30 patients (64 %) et de nouvelles anomalies étaient retrouvées chez 17 patients (36 %). Les patients ayant des titres élevés d’aCL étaient plus à risque de développer de nouvelles lésions cardiaques.
II.2. Manifestations coronariennes
Les atteintes coronaires observées au cours du SAPL relèvent de deux mécanismes principaux la thrombose et l’athérosclérose accélérée. Dans une étude portant sur 4081 hommes sains d’âge moyen, Vaarala et al avaient montré que la présence d’un titre élevé d’aCL était un facteur de risque indépendant de survenue d’infarctus du myocarde (IDM) ou de décès d’origine cardiaque. Ce risque relatif était de l’ordre de 2,0. C’est un risque indépendant des facteurs confondants comme l’âge, le tabac, la pression artérielle, le taux de LDL ou de HDL. La prévalence des aPL chez des patients faisant un IDM est de l’ordre de 5 à 15 % [56]. La persistance d’un titre élevé d’aCL constitue même un facteur de risque indépendant de récidive d’événement cardiaque conférant un risque
relatif équivalent à celui d’un tabagisme actif ou d’un diabète [57]
. Si la recherche systématique d’aPL chez tout patient faisant un IDM n’est pas indiquée, en revanche, elle est nécessaire dans les conditions suivantes : sujet âgé de plus 45 ans, antécédents de thrombose artérielle ou veineuse ou de perte fœtale à répétition, et sujets ayant des antécédents familiaux de maladies auto-immunes, en particulier le lupus.
En outre, il a été retrouvé une corrélation entre le titre des aCL et les anticorps anti-LDL oxydés. Les anticorps anti-LDL oxydés sont considérés comme des marqueurs d’athérosclérose. La présence d’aCL et d’anticorps anti-LDL oxydés semble conférer à ces deux anticorps un risque additif d’IDM. La présence d’anticorps anti-LDL oxydées a pu être confirmée au cours du SAPL primaire ou secondaire au LES soulignant l’importance des phénomènes d’athérosclérose chez ces patients. Ces phénomènes d’athérosclérose sont vraisemblablement secondaires à la réaction croisée entre aCL et anti-LDL oxydées, à l’internalisation des LDL oxydées en présence d’anticorps anti- 2
-GPI et aux immuns complexes formés qui sont, eux aussi, internalisés par les macrophages dans la paroi endothéliale, aggravant le développement de la plaque d’athérome. Par ailleurs, en particulier au cours du LES, des anticorps anti-HDL ont pu être mis en évidence [58]. Leur titre est inversement corrélé à l’activité de la paraoxonase, enzyme diminuant la peroxydation lipidique et donc la formation de la plaque d’athérome. L’activité de cette enzyme est abaissée chez les patients porteurs d’aCL dans plusieurs études, qu’ils aient un SAPL primaire ou secondaire [58]. De plus, au cours du LES avec aPL, on note un profil proathérogène associant une baisse du HDL-cholestérol et de l’apolipoprotéine A1 qui est vraisemblablement lié à une activité anti-HDL et anti-ApoA1 des
aCL [59]. Une insuffisance coronarienne est aussi possible, elle peut précéder ou suivre la survenue d’un IDM [56]
. Un angor instable peut aussi être un mode de présentation du SAPL. Dans l’étude Euro-APS portant sur 1000 sujets, les signes d’angor étaient retrouvés chez 2,7 % des patients [52]
.
II.3. Hypertrophie et dysfonction ventriculaire
Les données concernant le SAPL et la fonction ventriculaire sont peu nombreuses.
II.3.1. Fonction ventriculaire droite
Tektonidou et al en 2001 [60] soulignent que les patients porteurs d’un SAPL primaire ou secondaire au LES ont une altération significative de la fonction diastolique du ventricule droit, en particulier pour le SAPL primaire. De plus, l’ancienneté du SAPL, la présence d’une hypertension pulmonaire, le titre d’aCL IgG sont corrélés positivement à l’altération de la fonction diastolique du ventricule droit.
II.3.2. Fonction ventriculaire gauche
Au cours du SAPL primaire, il existe aussi des anomalies de la fonction diastolique du ventricule gauche, ainsi que de son remplissage. Cependant, ces résultats doivent être considérés avec prudence, d’autres facteurs confondants existent que ce soit les valvulopathies associées ou l’ischémie coronarienne.
On a décrit au cours du SAPL primaire des thromboses de la microcirculation myocardique à l’origine de tableaux d’IDM à coronarographie normale. Ces thromboses du réseau microcirculatoire peuvent entraîner une
hypertrophie myocardique et une dysfonction myocardique. Bien entendu, les atteintes valvulaires sont aussi une cause possible d’hypertrophie et de dysfonction ventriculaire, de même que les accidents thromboemboliques peuvent être source de défaillance ventriculaire droite [1].
II.4. Thrombus intracardiaque
Au cours du SAPL, qu’il soit primaire ou secondaire au LES, des thrombi- intracardiaques peuvent se développer dans toutes les cavités cardiaques. C’est une manifestation rare, rapportée dans seulement 0,4 % dans l’étude Euro-APS
[52]
. Les thrombi intracardiaques sont habituellement retrouvés à l’occasion d’une complication embolique. Les cavités droites sont plus souvent concernées que les gauches, à l’inverse des atteintes valvulaires. Si les aPL contribuent à la formation de ces thrombi, le mécanisme précis de leur formation est peu clair. L’échographie transoesophagienne peut être mise en défaut car les thrombi des cavités droites sont parfois difficiles à bien repérer. En outre, il peut être impossible de les différencier du myxome, dans ce cas, l’imagerie par résonance magnétique (IRM) cardiaque est utile. Après injection de gadolinium, le myxome se rehausse en T2 habituellement, alors que le thrombus non [1].
II.5. Hypertension artérielle pulmonaire [1]
La fréquence de l’hypertension pulmonaire est estimée à 1,8 % du SAPL secondaire au LES et à 3,5 % du SAPL primaire. Cette fréquence est de 2,2 % dans l’étude Euro-APS [52]. Il s’agit le plus souvent d’une hypertension
d’une hypertension pulmonaire post-embolique varie entre 10 et 20 %. Au cours du LES, l’hypertension pulmonaire est significativement associée aux aPL [10]
. De plus, ces aPL ne semblent pas être simplement satellites de l’hypertension pulmonaire comme le suggère leur fréquence bien plus élevée en présence d’une hypertension pulmonaire post-embolique qu’en présence d’une HTAP idiopathique. Plus rarement, une hypertension artérielle portopulmonaire ou une hypertension pulmonaire par maladie veino-occlusive peut aussi survenir chez des patients ayant des aPL [61]. Le sombre pronostic des hypertensions pulmonaires postemboliques observées au cours du SAPL nécessite une forte anticoagulation (INR ≥ 3).
III. COMPLICATIONS OBSTETRICALES [62]
Les complications obstétricales semblent être majoritairement la conséquence de thromboses placentaires ou fœtales, bien que d’autres mécanismes soient aujourd’hui discutés. Il s’agit en premier lieu des pertes fœtales précoces (<10 SA ou fausses couches spontanées) ou tardives (>10 SA ou morts fœtales in utero), des problèmes d’infertilité, de la prééclampsie, des thromboses veineuses ou artérielles chez la mère (incluant les AVC), des complications liées au traitement. Chez les patientes ayant un LES associé, on peut également observer des poussées de la maladie.
Infertilité :
Plusieurs travaux ont montré que la stérilité primaire était associée à une prévalence plus élevée d’aPL, estimée selon les études de 24 % à 42 %. En plus d’interférer avec la croissance fœtale, les aPL pourraient en effet jouer un rôle
sur la nidation placentaire. Ainsi, il a été montré in vitro, que la β2-GPI se fixe via un domaine particulier au trophoblaste et est reconnue par les anticorps anti-β2-GPI. Cette liaison antigène-anticorps entraînerait une diminution de la synthèse et de la sécrétion d’Human Chorionic Gonadotropin (HCG). Ces données pourraient expliquer les difficultés de placentation liées à la présence d’aPL de même que les pertes fœtales.
Fausses couches précoces (< 10 SA) et morts fœtales (>10 SA) :
Les fausses couches précoces (FCP) sporadiques sont fréquentes dans la population générale. En revanche, leur caractère récurrent (au moins 3 épisodes) est beaucoup plus rare, et parmi quelques autres étiologies (génétiques, hormonales, anomalies utérines), les aPL jouent un rôle incontestable. Une étude a ainsi montré, chez 366 femmes ayant eu au moins 2 épisodes de pertes fœtales, que 76 (21 %) d’entre elles avaient soit des aCL, soit un ACC, soit les deux. Parmi ces 76 femmes avec aPL, 50 % de ces pertes fœtales survenaient après 10 SA, contre seulement 10 % chez les patientes sans aPL. Dans une population de patientes ayant un SAPL connu, le pourcentage de pertes fœtales précoces, au cours du 1er trimestre, varie de 25 à 67 %.
Prééclampsie, retard de croissance intra-utérin, prématurité
Dans les différentes séries de grossesses menées chez des patientes avec SAPL, une hypertension artérielle gestationnelle voire une prééclampsie est observée dans 32 % des cas, pouvant aller jusqu’à 50 % des cas. Les insuffisances placentaires avec retard de croissance intra-utérin et justifiant une mise au monde de l’enfant sont également fréquentes, rapportées dans quelques
unes de ces séries. Il est donc peu surprenant d’observer un taux de naissances prématurées dans ces mêmes séries allant de 32 à 65 %. Paradoxalement et par opposition au fort taux de prééclampsie survenant chez les patientes ayant un SAPL, on ne retrouve pas plus d’immunologie antiphospholipide dans la population des femmes qui ont une prééclampsie, ni chez les patientes à risque de prééclampsie (antécédent de prééclampsie lors d’une précédente grossesse, hypertension artérielle chronique).
IV. SYNDROME CATASTROPHIQUE DES ANTIPHOSPHOLIPIDES
Une évolution grave et potentiellement létale du SAPL a été décrite pour la première fois par Asherson et dénommée syndrome catastrophique des aPL (SCAPL) [63].
Il justifie d’être individualisé en raison de ses particularités physiopathologiques et de sa gravité, avec une mortalité voisine de 50 % [64]. Des critères préliminaires de classification de SCAPL ont été rapportés lors du Xème congrès international sur les antiphospholipides en 2002 à Taormina. Ces critères sont rappelés par Erkan et al [65] (Tableau VI).
Il est schématiquement défini par la survenue d’une atteinte ischémique touchant au moins trois organes, s’installant en moins d’une semaine avec la confirmation histopathologique d’une oblitération des petits vaisseaux, associées au plan biologique à la présence d’aPL qui sont habituellement présents à un titre élevé. Il est lié à des phénomènes d’oblitérations thrombotiques au niveau de la microcirculation, ce qui le rapproche des microangiopathies thrombotiques
telles que le syndrome hémolytique et urémique, le HELLP syndrome [66] ou le syndrome de Moschcowitz, dont il partage d’ailleurs de nombreuses manifestations cliniques et biologiques. Il existe en particulier fréquemment une thrombopénie et une hémolyse mécanique avec présence de schizocytes. Un déficit de l’activité protéase ADAMTS-13 a occasionnellement été rapporté chez des patients atteints d’un SAPL dans sa forme catastrophique [67]
.
En revanche, l’activité ADAMTS-13 est normale chez des patients atteints d’un SAPL dans une forme non catastrophique et les aPL sont habituellement absents chez des malades atteints d’un syndrome de Moschcowitz. Le SCAPL peut révéler un SAPL méconnu jusqu’ici et il peut survenir aussi bien au cours du SAPL primaire qu’au cours du lupus. Un facteur déclenchant est souvent retrouvé, tels une infection, un traumatisme, une intervention chirurgicale ou l’arrêt récent d’un traitement anticoagulant. Cela souligne les dangers de la chirurgie chez les patients atteints de SAPL et les mesures de préventions rigoureuses qui doivent l’accompagner, avec en particulier la nécessité d’assurer une parfaite gestion de l’anticoagulation prophylactique sur ce terrain. Cela illustre également la difficulté de poser l’indication de l’arrêt d’un traitement anticoagulant après un premier épisode thrombotique chez un patient atteint de SAPL en raison du haut risque de récidive de thrombose, éventuellement sous la forme d’un SAPL catastrophique. Le SAPL catastrophique se manifeste par un tableau de défaillance multiviscérale qui peut comporter une atteinte cardiaque souvent grave qui représente la principale cause de décès, une atteinte respiratoire sous la forme d’un syndrome de détresse respiratoire aiguë pouvant être associé à une hémorragie intra-alvéolaire, une HTA maligne avec insuffisance rénale, un infarctus splénique et une hémolyse avec présence de
schizocytes, des signes neurologiques centraux pouvant entraîner un état de mal convulsif.
Au plan biologique, on note parfois des signes de coagulation intravasculaire disséminée. L’existence d’une atteinte rénale, splénique ou pulmonaire a une valeur pronostique péjorative. Le pronostic serait également plus grave en cas de lupus.
Le SAPL dans sa forme catastrophique mérite clairement d’être connu des hématologistes car si le tableau clinique et biologique peut être proche de celui du syndrome de Moschcowitz, la prise en charge thérapeutique, qui sera détaillée ultérieurement, est différente, avec notamment la nécessité d’utiliser une héparinothérapie et des corticoïdes alors que la place des échanges plasmatiques reste très discutée dans cette situation [68].
Tableau VI : Critères préliminaires de classification du SCAPL [65]. Critères
1. Mise en évidence de l’atteinte d’au moins 3 organes, systèmes et/ou tissus (a) 2. Survenue simultanée des différentes atteintes ou en moins d’une semaine
3. Confirmation histologique de l’occlusion des petits vaisseaux dans au moins un organe ou tissu (b)
4. Confirmation biologique de la présence d’aPL (LA et/ou aCL et/ou anticorps anti- 2-GPI) (c)
Syndrome catastrophique des antiphospholipides défini
→Les 4 critères sont rassemblés
Syndrome catastrophique des antiphospholipides probable
→Les 4 critères sont rassemblés, mais seulement 2 organes, systèmes et/ou tissus sont concernés
→Les 4 critères sont rassemblés, mais la confirmation biologique de la persistance à 6 semaines des aPL n’a pu être réalisée du fait de la mort précoce du patient ou de l’absence de test avant la survenue du SCAPL
→1, 2 et 4
→1, 3 et 4 et survenue d’un 3ème événement plus de 1 semaine, mais moins de 1 mois, avant les premières manifestations, malgré le traitement anticoagulant.
(a) : Mise en évidence clinique d’occlusion vasculaire, confirmée par des techniques d’imagerie appropriées. L’atteinte rénale se définit comme une augmentation d’au moins 50 % du taux de la créatininémie, l’apparition d’une hypertension artérielle sévère (> 180/100mmHg) et/ou d’une protéinurie (> 500 mg/24 h).
(b) : Confirmation histologique signifie la mise en évidence d’un phénomène de thrombose bien qu’un processus de vascularite puisse occasionnellement coexister.
(c): Si le patient n’a jamais eu de test biologique au préalable, les anticorps antiphospholipides doivent être confirmés à au moins deux reprises, espacées d’au moins 6 semaines (pas nécessairement au moment de l’événement clinique), en accord avec les critères préliminaires proposés pour la classification des SAPL de Sapporo.
V. SAPL ET CANCER
L’association cancer et aPL a été décrite dans un nombre important d’études épidémiologiques. La présence d’aPL augmente le risque thrombotique, la thrombose étant bien connue comme complication possible dans l’évolution d’un cancer [69, 70]
. Trousseau en 1865 a été le premier à signaler la corrélation de la néoplasie avec les évènements thrombotiques. Dans une étude sur 216 malades avec tumeurs solides, Zuckerman [71] met en évidence la présence d’aPL chez 21,8 % des malades. Parmi ce dernier groupe, 27,7 % présentent des complications thrombotiques. Gomez-Puerta [72] sur une période comprise de 1996 à 2003 a diagnostiqué 120 malades présentant un cancer associé aux aPL. Dans 71,7 % des cas, il avait diagnostiqué la présence de complications thrombotiques dont 21,7 % remplissaient les critères de Sapporo. Une hétérogénéité de tumeurs solides a été identifiée, parmi lesquelles il y avait une prédominance d’adénocarcinomes pulmonaire, ovarien, rénal et de la prostate. Ces complications thrombotiques sont plus fréquentes chez les malades
avec tumeurs solides : 46 %, qu’en pathologie hématologique maligne : 32 %
[73]
. Dans une étude asiatique une prévalence importante des anti-β2 immunoglobulines A a été mise en évidence chez les malades avec cancer et thromboses [74]. Le mécanisme d’apparition des aPL en pathologie maligne n’est pas élucidé. Une multitude des facteurs a été incriminé comme suit : la libération du tissu facteur du « cancer procoagulant » ou même la dérégulation du lymphocyte B. Les aPL peuvent être considérés «épiphénomènes» de la maladie maligne, c’est le cas des maladies hématologiques malignes [75]
. Dans ce cas leur présence est liée à la synthèse d’une gammapathie monoclonale rarement responsable d’une thrombose. En revanche, chez les malades avec tumeurs solides ou l’incidence de thromboses est plus élevée, les aPL peuvent avoir une implication pathogénique dans les mécanismes de la thrombose. Les aPL ne développent pas de manière systématique des complications thrombotiques sans un facteur initiateur d’origine inflammatoire, infectieuse ou néoplasique. Cette constatation qui est à l’origine du concept « two-hit » suggère que chez les malades cancéreux les aPL interviendraient en second lieu d’un état d’hypercoagulabilité induit par la pathologie maligne.
Différentes circonstances cliniques interviennent dans l’évolution des taux d’aPL. Les aPL peuvent augmenter dramatiquement après une intervention chirurgicale sur la tumeur ou même après une biopsie [76].
En revanche, les taux d’aPL diminuent et même disparaissent après une chimiothérapie efficace dans un cancer du côlon. De même leurs taux se normalisent chez 40 % des lymphomes après chimiothérapie, mais remontent en période de rechute [77]. Cause ou conséquence de la malignité, l’implication des aPL n’est pas encore clarifiée. Lors d’une étude rétrospective portant sur des cas
considérés comme étant « des thromboses paranéoplasiques » Asherson conclut que ces malades remplissaient en réalité les critères d’un SAPL.
Malheureusement, à l’heure actuelle aucune étude prospective n’a évalué, à long terme et en cinétique, la corrélation entre le taux d’aPL et l’évolution clinique de la maladie maligne après chimiothérapie, exérèse chirurgicale, rémission ou rechute [78].
VI. SAPL ET LUPUS [79]
C’est au cours du lupus que les aPL ont été décrits pour la première fois il y a 50 ans avec la mise en évidence de l’anticoagulant circulant lupique. Depuis une vingtaine d’année, l’individualisation du SAPL a profondément modifié la prise en charge du lupus en permettant de mieux comprendre la physiopathologie de certaines complications viscérales, notamment neurologiques, cardiovasculaires ou rénales. Il est important de différencier, devant une complication survenant au cours du lupus, ce qui revient à une thrombose satellite du SAPL et à une complication immunologique plus spécifique du lupus. La recherche d’aPL doit faire partie du bilan biologique systématique réalisé lorsqu’un diagnostic de lupus est porté. Il a même été proposé que la présence d’aCL fasse désormais partie des critères diagnostiques de lupus. La présence d’aPL est trouvée chez environ 30 % des patients lupiques. En l’absence de prophylaxie, le risque d’accident thrombotique serait de 50 %. Les travaux initiaux suggéraient que la présence d’aPL avait une influence péjorative sur le pronostic du lupus. Cette donnée est contestée par les études les plus récentes, probablement en raison d’une meilleure prévention des accidents thrombotiques. Les travaux de Petri et al non encore publiés mais
présentés récemment au 10th international congress on antiphospholipid antibodies en Italie notent, cependant une grande fréquence d’infarctus du myocarde (21%) et d’évènements cardiovasculaires (19 %) au sein d’une cohorte de 1000 patients lupiques suivis aux Etats-Unis. Cette fréquence d’événements cardiovasculaires est significativement plus élevée chez les patients lupiques ayant un ACC. Lorsque le SAPL est isolé au départ, il est exceptionnel qu’il évolue ultérieurement vers un lupus. En revanche, certains patients répondant initialement aux critères diagnostiques de SAPL ont des signes de lupus sans remplir pour autant tous les critères de l’American College of Rheumatology. Il a été proposé le terme de lupus-like syndrome pour caractériser cette situation. Piette et al ont proposé une série de signes permettant, dans cette situation, d’éliminer le diagnostic de SAPL primaire. Parmi eux, on retiendra principalement la présence d’anticorps antinucléaires à un titre supérieur à 1/320.
VII. COMPLICATIONS OSTEO-ARTICULAIRES
Il s’agit d’ostéonécroses aseptiques épiphysaires surtout fémorales qui compliquent surtout le SAPL associé au lupus. Ces complications sont très rares dans le SAPL primaire, ce qui suggère le rôle de facteur associé comme les corticoïdes, souvent utilisés dans le lupus. Plus rarement, il s’agit de nécrose osseuse plus étendue [80].
VIII. MANIFESTATIONS CUTANEES [81]
Les manifestations cutanées incluent le livedo reticularis (Fig. 4), parfois associé à des AVC dans le cadre d’un syndrome de Sneddon (40 % comportent des aPL), des ulcérations cutanées, une pseudovascularite (macules
érythémateuses, purpura douloureux, bulles hémorragiques), des nécroses cutanées superficielles, une gangrène digitale (Fig. 5), des hémorragies en «flammèche sous-unguéales» (Fig. 6), une papulose atrophiante maligne de Degos.
Figure 4: Livedo réticulaire des membres inférieurs au cours du SAPL [81].
Figure 5: Gangrène distale digitale [81]. Figure 6: Hémorragies
IX. SAPL ET SYSTEME NERVEUX
Les liens entre aPL et système nerveux ont fait l’objet de nombreuses publications mais de nombreux points importants restent en suspens [82].
La manifestation neurologique la plus fréquente et pouvant révéler le SAPL est la survenue d’un accident vasculaire cérébral ischémique constitué ou transitoire par thrombose artérielle ou par embolie à point de départ cardiaque
[83]
.
La survenue d’un tel accident chez un sujet jeune doit faire rechercher un SAPL. Une telle recherche chez un sujet âgé porteur de facteurs de risque athéromateux serait abusive. L’association d’un accident neurologique ischémique à un livédo définit le syndrome de Sneddon. Des aPL y sont présents dans 40 % des cas [84]. Les thrombophlébites cérébrales sont plus rares. Les liens entre aPL et sclérose en plaque (SEP) sont discutés et certaines équipes ont rapporté des tableaux neurologiques proches de celui de la SEP chez des patients porteurs d’aPL [85]
. Il existe néanmoins des nuances sémiologiques cliniques et iconographiques qui permettent habituellement de trancher entre ces deux étiologies [86]. On retiendra principalement un mode de début brutal pour le SAPL, la présence de signes d’appel extra-neurologiques, l’atteinte neurologique correspondant à des territoires artériels ou veineux, la possibilité d’une atteinte de la substance grise à l’IRM avec des lésions moins étendues qu’au cours de la SEP et touchant plus volontiers les ganglions de la base. Il existe cependant des formes frontières entre ces deux entités pour lesquelles il est difficile de trancher. Cette discussion n’est pas uniquement académique car cette distinction a des conséquences thérapeutiques importantes [85]. D’autres
manifestations neurologiques peuvent être associées au SAPL comme les troubles des fonctions supérieures, la chorée, les myélites transverses et peut être certaines formes de migraine [87].
X. SAPL ET THROMBOPENIES [19]
Schématiquement, 2 types de thrombopénies dont le mécanisme et les conséquences sont différents peuvent être rencontrés dans le SAPL : une thrombopénie par destruction plaquettaire accélérée de mécanisme immunologique (auto-anticorps concomitants aux aPL, et dirigés spécifiquement contre les glycoprotéines majeures et spécifiques des plaquettes) peut entraîner un risque hémorragique comme au cours de toute thrombopénie auto-immune, alors qu’une thrombopénie conséquence d’une activation de la coagulation (via la thrombine) et d’un processus de consommation va être associé à un risque de thrombose. Il peut donc être difficile face à un patient à un moment donné de déterminer le mécanisme et la signification de la thrombopénie si l’on ne dispose pas d’éléments évolutifs. Ceci explique que la thrombopénie ne fasse plus partie des critères de classification de Sydney comme cela avait été le cas des premières tentatives de classification. Pour une thrombopénie associée aux antiphospholipides, la persistance du risque thrombotique justifie de poursuivre le traitement anticoagulant si cette indication a été portée en raison d’antécédents de thrombose (du moins si le compte plaquettaire est > 50 G/L). Il est cependant possible que la thrombopénie soit un facteur protecteur des récidives thrombotiques. Si la thrombopénie est grave et est associée à un syndrome hémorragique, le traitement immunomodulateur ne diffère pas de celui des autres thrombopénies auto-immunes. Il faut cependant être conscient
![Tableau II : Les phospholipides cibles des aPL [13] .](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/9.892.151.744.172.474/tableau-ii-phospholipides-cibles-des-apl.webp)
![Tableau III : Critères de classification révisés du syndrome des aPL (critères de Sydney) [19] .](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/12.892.98.789.193.750/tableau-iii-critères-classification-révisés-syndrome-critères-sydney.webp)
![Figure 2 : Activation de la coagulation par le TF associé aux phospholipides de membrane en cas d’absence d’annexine V [27]](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/21.892.117.789.105.706/figure-activation-coagulation-associé-phospholipides-membrane-absence-annexine.webp)
![Figure 3 : Rôle de l’activation du complément dans les pertes fœtales induites par les aPL [27]](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/25.892.106.777.97.670/figure-rôle-activation-complément-pertes-fœtales-induites-apl.webp)
![Figure 4: Livedo réticulaire des membres inférieurs au cours du SAPL [81 ] .](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/47.892.254.641.271.536/figure-livedo-réticulaire-membres-inférieurs-cours-sapl.webp)
![Tableau VII : Critères de positivité des tests de diagnostic des LA [108]](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/65.892.111.788.175.433/tableau-vii-critères-positivité-tests-diagnostic.webp)
![Tableau VIII : Diagnostic différentiel entre LA, anticorps anti-facteur de la voie endogène et déficit en facteur [108]](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/68.892.103.836.199.641/tableau-diagnostic-différentiel-anticorps-facteur-endogène-déficit-facteur.webp)
![Figure 7 : Arbre décisionnel. Approche schématique pour l’exploration biologique du SAPL [151]](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/88.892.208.687.109.619/figure-arbre-décisionnel-approche-schématique-exploration-biologique-sapl.webp)
![Tableau XI: Quelques séries cliniques randomisées publiées [181] Etudes/Année Nombre de patientes 1 er bras de traitement 2 e bras de traitement Naissances vivantes 1er bras Naissances vivantes 2ebras Commentaires Cowchock, 1992 [182] 20](https://thumb-eu.123doks.com/thumbv2/123doknet/14392399.701370/101.892.59.835.177.931/cliniques-randomisées-publiées-traitement-traitement-naissances-naissances-commentaires.webp)
