P
Prréésseennttééppaarr::
Docteur Sara JOULAIBI
E
Ennccaaddrrééppaarr::
Professeur Mounia RAHMANI
Septembre 2020
ROYAUME DU MAROC
Université Mohammed V - Rabat Faculté de Médecine et de Pharmacie
RABAT
M
M
é
é
m
m
o
o
i
i
r
r
e
e
d
d
e
e
f
f
i
i
n
n
d
d
’
’
é
é
t
t
u
u
d
d
e
e
s
s
Pour L’obtention du Diplôme National de Spécialité
en
NEUROLOGIE
Intitulé
L
L
A
A
P
P
A
A
R
R
A
A
L
L
Y
Y
S
S
I
I
E
E
S
S
U
U
P
P
R
R
A
A
N
N
U
U
C
C
L
L
E
E
A
A
I
I
R
R
E
E
P
P
R
R
O
O
G
G
R
R
E
E
S
S
S
S
I
I
V
V
E
E
:
:
A
A
p
p
r
r
o
o
p
p
o
o
s
s
d
d
e
e
1
1
2
2
c
c
a
a
s
s
Sommaire
I-Introduction ... 2
II- Rappel sur la paralysie supranucléaire progressive ... 5
A. Données anatomopathologiques ... 5
B. Epidémiologie ... 8
C. Symptomatologie clinique ... 8
a. Troubles oculomoteurs ... 10
b. Troubles posturaux et chutes ... 11
c. Akinésie ... 11
d. Troubles cognitifs et comportementaux ... 12
D. Formes cliniques ... 13
a. Syndrome de Richardson ... 13
b. La paralysie supranucléaire progressive-parkinsonisme ... 14
c. PSP- Freezing ... 14
d. PSP-Syndrome cortico-basal ... 15
e. PSP-Frontale ... 15
f. PSP-Aphasie non fluente ... 16
g. PSP- syndrome cérébelleux ... 16
E. Examens complémentaires ... 18
a. Imagerie cérébrale ... 18
F. Critères diagnostiques ... 24
G. Traitement ... 29
III- Matériel et méthodes ... 34
1.Type d’étude ... 34
2. Critères d’inclusion et d’exclusion ... 34
3. Collecte des données ... 35
IV- Résultats ... 40 A. Observations cliniques ... 40 1. Mr B. S ... 40 2. Mme F. B ... 40 3. Mme B. F ... 41 4. Mr C. E ... 41 5. Mr E. B ... 42 6. Mr E. A ... 42 7. Mr E. M ... 43 8. Mr H. M ... 44 9. Mr I. M ... 44 10. Mme N. E ... 45 11. Mr O. E ... 45 12. Mr T. A ... 46
B. Analyses des données ... 46
I. Description des patients ... 46
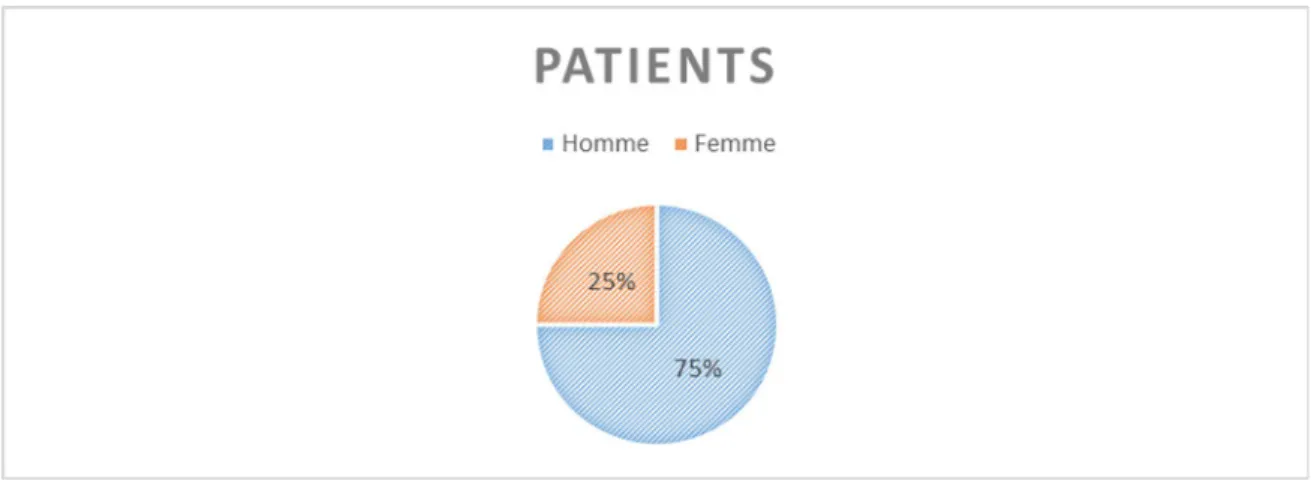
2. Sexe ... 46
3. Antécédents médicaux-chirurgicaux ... 47
II. Étude clinique ... 48
1. Signes de début ... 48 2. Autres symptômes ... 49 3. Formes cliniques ... 50 4. Examens paracliniques ... 51 a. IRM cérébrale ... 51 b. Bilan biologique ... 51 5. Traitement ... 52 6. Evolution et suivi ... 52 V. Discussion ... 54 Conclusion ... 62 Résumé ... 64 Références ... 66
Liste des figures
Figure 1: Epissage des différents isoformes de la protéine tau ... 7
Figure 2: Caractéristiques histopathologiques principales de la PSP ... 7
Figure 3: Caractéristiques cliniques des différentes formes cliniques de PSP . 17 Figure 4: Principaux signes cliniques distinctifs des différentes formes de PSP avec la maladie de parkinson ... 18
Figure 5: A. Coupe axiale T1 du mésencéphale : Aspect en «oreilles de Mickey» et en «fleur de liseron» (flèches). B. Coupe sagittale du tronc cérébral : Aspect de bec d’oiseau : « Signe du pingouin». ... 20
Figure 6: signe de colibri, objectivé par l’aspect du bec d’oiseau visible sur une coupe IRM sagittale T1 (image B) ... 20
Figure 7: IRM cérébrale en coupes sagittales T2 d’un patient souffrant d’une PSP, objectivant une atrophie du mésencéphale (signe de colibri) et l’atrophie frontale nettement visibles avec l’évolution de la maladie ... 21
Figure 8: TEP cérébrale au 18FDG chez une patiente de 65 ans atteinte de PSP-P. Notez l'hypométabolisme fronto-cingulaire mésial et orbitofrontal et la relative préservation des aires postérieures et des ganglions de la base. À noter qu'il existe ici également une discrète atteinte pariétale supérieure bilatérale habituellement retrouvée dans les syndromes cortico-basaux. ... 23
Figure 9: Conduite à tenir devant une suspicion d’une PSP ... 27
Figure 10: Répartition des patients selon le sexe ... 47
Figure 11: Répartition des antécédents médico-chirurgicaux ... 48
Figure 12: Répartition des symptômes cliniques de début de la maladie. ... 49
Figure 13: Répartition des formes cliniques ... 50
Liste des tableaux
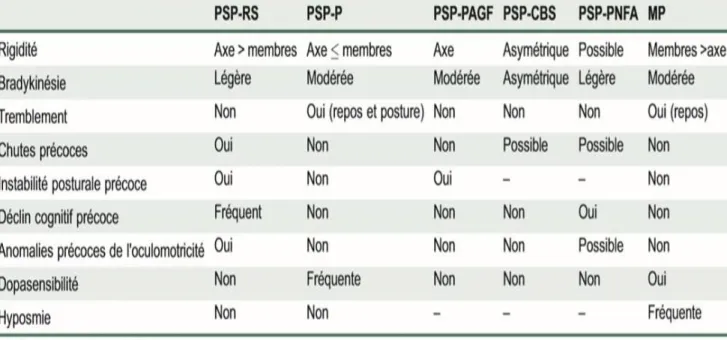
Tableau 1: Niveaux de certitude du diagnostic de PSP en fonction des signes fonctionnels formant le noyau clinique de la maladie. ... 28
Tableau 2: Signes cliniques et IRM associés à la PSP ... 28 Tableau 3: Critères diagnostiques des différentes formes de la PSP selon les symptômes fonctionnels et anomalies IRM ... 29
I-Introduction :
La PSP est une affection sporadique neurodégénérative de l’adulte, caractérisée sur le plan neuropathologie par l'accumulation de protéine tau hyperphosphorylée dans les ganglions de la base et le tronc cérébral (1).
La 1ère description d’une personne souffrant d’une forme classique de PSP était celle de l’écrivain romancier Charles Dickens en 1857 dans son roman intitulé : ‘The lazy tour of two idle apprentices’ (2) : « Un vieil homme froid, lent, terreux et fixe. Un homme cadavérique au discours restreint. Un vieil homme qui semblait incapable de cligner de l'œil, comme si ses paupières avaient été clouées sur son front. Un vieil homme dont les yeux, deux taches de feu, n'avaient plus de mouvements comme s'ils avaient été reliés à l'arrière de son crâne par des vices (…). Il était entré et avait fermé la porte, et il s'assit maintenant. Il ne s'est pas penché pour s'asseoir, comme le font les autres, mais a semblé s'enfoncer tout droit, comme s'il était dans l'eau, jusqu'à ce que la chaise l'arrête. »
Le 1er scientifique à rapporter un cas de PSP est le neurologue canadien Richardson, qui décrivit la maladie en 1963 comme étant une association d’une rigidité axiale, d’une instabilité posturale, d’une paralysie supranucléaire, de troubles cognitifs et d’un syndrome pseudo bulbaire (3). Ses collaborateurs, Jerzy Olsewzky et John Steele, l’ont dénommée par la suite comme une nouvelle entité nosologique : la paralysie supranucléaire progressive.
Les critères neuropathologiques de la PSP ont été posés dans les années quatre-vingt-dix (4, 5). Il est actuellement évident que le phénotype initialement décrit, actuellement appelé syndrome de Richardson (PSP-RS), n’est que l’un des nombreux phénotypes cliniques de la PSP. Steele (6) a noté que des variantes cliniques du syndrome sont susceptibles de se produire car la maladie affecte différents noyaux à différents moments et à différents degrés. Même si toutes les formes cliniques partagent des caractéristiques neuropathologiques
similaires remplissant les critères diagnostiques de la PSP, la reconnaissance croissante de l'hétérogénéité phénotypique est liée à l’importance d'accumulation anormale de la protéine tau et de la perte neuronale dans certaines régions particulières du cerveau (7). Cette accumulation se manifeste comme une dégénérescence neurofibrillaire sous-corticale étendue trouvée principalement dans le globus pallidus, le noyau subthalamique, la substance noire et le noyau dentelé cérébelleux. L'hétérogénéité de la présentation clinique est reconnue dans les critères diagnostiques actuels de la PSP mis à jour par la MDS en 2017 (5).
Nous présentons une série rétrospective de malades ayant une PSP, suivis au service de Neurologie A de l’hôpital des spécialités entre 2015 et 2020. L’analyse de cette série a pour but de décrire le profil clinique, les variantes phénotypiques, les données de l’IRM ainsi que le profil évolutif de notre population. Les résultats sont discutés et comparés aux données de la littérature.
Rappel sur la paralysie
supranucléaire
II- Rappel sur la paralysie supranucléaire progressive :
A / Données anatomopathologiques :
La PSP est la tauopathie primaire la plus courante appartenant à la famille des tauopathies 4R, correspondant à l'accumulation de l'isoforme tau avec quatre répétitions dans le domaine de liaison microtubulaire (8). Les anomalies histologiques de cette affection sont : la perte neuronale, la gliose astrocytaire et l’accumulation neuronale et oligodendrogliale de protéine tau anormalement phosphorylée. Ces accumulations prennent la forme de dégénérescence neurofibrillaire, de fibres tortueuses et de touffes astrocytaires caractéristiques de la maladie (9). Les lésions se situent au niveau des noyaux gris de la base (le pons, la substance noire, le noyau sousthalamique et le pallidum), le colliculus supérieur, le tronc cérébral, les noyaux oculomoteurs, le cortex prémoteur, le cervelet et la substance grise périaqueducale de la moelle épinière. La PSP fait donc partie du groupe des tauopathies incluant la maladie d’Alzheimer, la dégénérescence cortico-basale (DCB), la démence fronto-temporale associée au parkinsonisme liée au chromosome 17 et la maladie de Niemann-pick de type C.
La protéine tau appartient à la famille des protéines associées aux microtubules et est exprimée principalement dans les neurones, et à moindre degré dans d’autres tissus non neuronaux (10). Cette protéine est impliquée dans la stabilisation et l’assemblage aux microtubules. Dans le cerveau humain adulte, six isoformes de la protéine tau sont produits par l’épissage alternatif de l’ARNm d’un seul gène localisé sur le chromosome 17q21 (11). Ces isoformes diffèrent par la présence ou non de séquences de 29 acides aminés (N1) et/ ou de 59 acides aminés (N2) sur l’extrémité N terminale, et par le nombre de répétition d’une séquence de 31 acides aminés dans le domaine de transport des microtubules : 3 répétitions (3R) ou 4 répétitions (4R) selon l’épissage alternatif de l’exon 10 (figure 6). Dans un cerveau humain adulte, les isoformes 3R et 4R sont en proportion similaire. La quantité relative de chaque isoforme varie selon
L’hétérogénéité des anomalies neuropathologiques rend compte des différents phénotypes cliniques. Les patients souffrant d’une PSP avec un syndrome parkinsonien (PSP-P) ont une tauopathie moins sévère que dans le syndrome de Steel Richardson (SR) (11, 12). La substance noire (SN) et le noyau sous thalamique (NST) sont les régions les plus sévèrement atteintes dans la PSP-P. Les signes cliniques qui différentient la PSP-P et du SR pourraient être liés à la déplétion dopaminergique moins sévère dans les régions extranigriques dans la PSP-P (cortex cérébral, pont, noyau caudé, noyau dentelé et substance blanche cérébelleuse) (13).
La distribution des anomalies histologiques des patients souffrant d’une PSP avec une aphasie progressive non fluente (PSP-APNF) est limitée à une atteinte pallido-nigro-luysienne (12). Ces données sont confirmées sur les études d’imagerie fonctionnelle montrant une réduction du métabolisme du glucose uniquement dans le mésencéphale avec une préservation du métabolisme frontal (14). La distribution restreinte de ces anomalies neuropathologiques explique la différence phénotypique et par conséquent le meilleur pronostic de cette forme particulière. Chez les patients PSP-APNF, la pathologie tau est plus sévère dans le gyrus frontal supérieur et dans le cortex temporal, mais l’atteinte est moins sévère dans le tronc cérébral et la SG sous corticale (9). La distribution des anomalies neuropathologiques dans la PSP-syndrome cortico basal (PSP-SCB) est similaire à celle rencontrée dans la DCB avec des lésions prédominant dans le cortex fronto-médian et pariétal inférieur (9, 15).
Figure 1: Epissage des différents isoformes de la protéine tau (15)
Figure 2: Caractéristiques histopathologiques principales de la PSP (16)
Examen immunohistochimique du striatum en post-mortem d’un patient ayant une PSP avec utilisation d’un anticorps anti protéine Tau phosphorylée (AD 2), montrant A : des agrégats cytosoliques (coiled bodies), B : touffe astrocytique, C : les neurofilaments, D : neurone (pré-enchevêtrement), E : l’enchevêtrement neurofibrillaire.
B. Epidémiologie :
La prévalence de la PSP est de 5,8 à 6,5 pour 100 000 habitants selon l’étude de Barrostini (1). Le taux d'incidence annuelle varie de 0,3 à 0,4 pour 100 000 habitants (17). L'incidence annuelle a augmenté ces dernières années à 1,1 pour 100 000 habitants, due probablement à une meilleure détermination des cas avec une reconnaissance plus approfondie de l’affection et de ses variantes cliniques (17). L'incidence annuelle augmente avec l'âge allant de 1,7 cas pour 100 000 habitants entre 50-59 ans à 14,7 pour 100 000 habitants entre 80-89 ans (18).
L'âge moyen de début de la maladie est d'environ 65 ans. Pratiquement aucun cas de paralysie supranucléaire progressive confirmée par l'autopsie n'a été rapporté chez des patients de moins de 40 ans (17). La prédominance est masculine avec un ratio d'environ 8 à 1. Cependant, dans d’autres études, il n’existe aucune prédominance (17).
C. Symptomatologie clinique :
La PSP est un syndrome parkinsonien atypique de type akinéto-rigide, d’installation insidieuse et symétrique chez la plupart des patients.
Une phase présymptomatique peut survenir chez des patients asymptomatiques mais qui peuvent éventuellement développer une authentique PSP par la suite. Actuellement, la phase présymptomatique ne peut être identifiée qu'en post mortem par la mise en évidence de variations histologiques typiques d’une PSP chez les patients cliniquement normaux. Etant donné que les nouveaux critères MDS PSP se concentrent sur le diagnostic et n'incluent pas la phase présymptomatique, cette dernière reste néanmoins considérée. Dans une série d'autopsies, une PSP ‘ infra clinique‘ était présente chez cinq des 233 individus (2,1%). Deux études similaires ont mis en évidence une PSP chez 4,6% de sujets âgés asymptomatiques et 4,6% des personnes de plus de 60 ans
dans une série d'autopsie japonaise. Ces chiffres sont en contraste frappant avec les faibles prévalences de la PSP-RS, ce qui suggère que la plupart des patients avec PSP ‘infraclinique’ ne développent pas de maladie manifeste. (19)
Le terme suggestif de PSP suggestive of PSP (soPSP) fait référence à la phase symptomatique précoce de la PSP avant que le tableau clinique ne soit au complet, dans laquelle quelques symptômes / signes cliniques sont clairement présents, mais ne justifiant pas à eux seuls le diagnostic d’une PSP. L’évolution d’une soPSP vers une PSP authentique est difficile à prédire, D’où l’intérêt d’avoir des biomarqueurs spécifiques pour La PSP pouvant contribuer à un diagnostic précoce. La catégorie soPSP comprend également les patients ayant développé un ou plusieurs signes majeurs de la PSP-RS, et non le RS complet, comme par exemple les patients présentant un ralentissement des saccades isolé ou une instabilité posturale. Un diagnostic de soPSP ne peut être retenu que chez des patients dont la présentation clinique évoque une PSP, mais ne répondant pas pleinement aux critères de PSP, et ce d’autant plus si les diagnostics différentiels sont éliminés. Le but d’identifier les patients au stade soPSP de la maladie, est d’initier de manière précoce un traitement neuroprotecteur pour stabiliser les patients avant l'apparition d'un handicap majeur (19).
Au début de la maladie, les anomalies motrices intéressent l’axe beaucoup plus que les membres. Les troubles de la marche et les chutes sont la manifestation initiale la plus courante. D'autres signes peu courants peuvent initier la maladie tels que : des sensations vertigineuses, un ralentissement moteur généralisé, un changement de personnalité et moins souvent un tremblement au repos (20). L'ophtalmoplégie supranucléaire est un signe marqueur de la PSP, qui peut se limiter initialement à un ralentissement des saccades verticales et n’être complète qu’à l’évolution tardive de la maladie.
Les différents modes de présentation et la variabilité clinique des tableaux ont conduit à des études évaluant l’hétérogénéité clinique et neuropathologique de la PSP. De nouvelles entités nosologiques ont ainsi été définies, confirmant le tableau décrit initialement par Richardson, mais conduisant aussi à la description de variantes phénotypiques. Ces dernières peuvent se distinguer en termes de sévérité, de variabilité des régions impliquées par le processus neuropathologique et par leurs caractéristiques cliniques. Néanmoins, l’histoire naturelle de ces différents phénotypes est semblable conduisant au décès en six à 12 ans (1).
a. Troubles oculomoteurs :
L'anomalie la plus caractéristique à l'examen de la PSP-RS classique est la présence d’une paralysie supranucléaire (PS) dans le regard vertical. La PS est également notée dans les autres phénotypes, témoignant du chevauchement que ceux-ci peuvent avoir avec la PSP-RS. Elle s'exprime cliniquement par la limitation des mouvements oculaires notamment dans le regard vertical, accompagnée d'une rétraction de la paupière. La présence d’un creusement du sillon du front en raison de l'hyperactivité frontale (signe du procerus) et d’un cou allongé donne une apparence faciale tout à fait caractéristique avec un regard « surpris » à ces patients, à la différence de l'hypomimie, au cours la maladie de Parkinson (MP). La présence de la PS verticale notamment vers le bas est caractéristique mais non spécifique de la PSP devant par conséquent faire alerter le clinicien (19).
D'autres anomalies oculomotrices ont été décrites au cours de la PSP telles que des secousses en onde carrée, une rétraction des paupières, une apraxie d’ouverture des paupières définie par l’incapacité à initier volontairement l'ouverture des paupières après une période de fermeture et la diminution de la cadence de clignotement (1).
b. Troubles posturaux et chutes :
L'instabilité posturale et les chutes sont parmi les manifestations cardinales de la PSP. A la marche on note un élargissement du polygone de sustention avec une tendance à la rétropulsion. L’instabilité posturale isolée est considérée comme un phénotype de la PSP d’autant plus si elle apparaît précocement (19).
Plus l'instabilité posturale apparaît tôt, plus sa valeur diagnostique est grande. Les nouveaux critères de PSP définissent trois stades de l'instabilité posturale, en fonction du degré de déficit dans les trois premières années après le début de la maladie. P1 est définie par des chutes non provoquées répétées dans les 3 ans suivant leur apparition, suite à une perte d'équilibre spontanée en position debout, ou la présence d’un antécédent de plus d'une chute non provoquée, dans les 3 ans suivant l'apparition des caractéristiques liées à la PSP. La tendance à tomber lors du test de poussé dans les 3 ans suivant le début définie le stade (P2). Le test de la poussé (pulse test) examine la réponse à une traction rapide et puissante sur les épaules avec l'examinateur debout derrière un patient debout, les yeux ouverts et les pieds confortablement écartés et parallèles, comme décrit dans l'article 3.12 du MDS-UPDRS. Enfin, plus de deux pas en arrière lors du test de la poussée dans les 3 ans avec un patient qui est toujours capable de récupérer son équilibre sans aide définie le stade P3 (2).
c. Akinésie :
Au cours de la forme PSP-P, certains patients peuvent présenter une rigidité asymétrique, avec un tremblement de repos typique et une réponse initiale à la lévodopa mimant ainsi une véritable MP. La plupart des patients présentent certains éléments du syndrome akinéto-rigide à savoir une rigidité axiale au niveau des membres supérieurs proximale aux épaules, tandis que la rigidité distale des membres et la bradykinésie peuvent être absentes. L'akinésie généralisée du corps entier est typique et souvent le corps n'est pas fléchi mais
ballant du corps, mais plutôt une marche d’allure cérébelleuse avec des troubles de l’équilibre en particulier vers l'arrière, ce qui ne peut être évident au début que lorsque le patient essaie de s'asseoir et de se lever d'une chaise basse. Le freezing est observé au cours de la forme PSP-PAGF. Cette forme est caractérisée par des blocages moteurs soudains et transitoires ou une hésitation à l’initiation de la marche, prédominant dans les 3 premières années de début, progressifs et non sensibles à la lévodopa. Une akinésie peut être présente, mais la rigidité des membres, le tremblement et la démence sont absents ou légers (2).
d. Troubles cognitifs et comportementaux :
Souvent, l’entourage familial rapporte chez les patients PSP un changement de personnalité tel qu’une apathie, un déficit de l’attention, une indifférence vis-à-vis des proches alors que les oublis viennent au second plan. Il existe aussi une lenteur du traitement des nouvelles informations, une perte de la capacité de prendre des initiatives, de prendre des décisions ou même de participer à une conversation avec des arguments, même pour des sujets banals. L’ensemble de ces symptômes évoque un tableau de démence sous corticale (1).
Le terme de « démence sous-corticale » a été introduit en 1974 par Albert, Feldman et Willis, pour décrire ce profil cognitif caractéristique de la forme PSP-RS. Ce syndrome est différent de celui observé dans les démences corticales caractéristiques de conditions telles que la maladie d'Alzheimer ou les démences frontotemporales. En effet, le tableau général des déficiences cognitives dans la PSP-RS correspond à un ralentissement cognitif, avec des difficultés particulières sur les tests visant à détecter des dysfonctionnements du lobe frontal. La mémoire, le langage et les praxis sont plutôt conservés dans la PSP-RS (19).
Cependant, il existe de nombreuses formes de la PSP qui peuvent avoir des caractéristiques cognitives prédominantes qui vont bien au-delà du profil classique de la PSP-RS (2).
D. Formes cliniques : (figures 3 et 4)
a. Syndrome de Richardson (PSP-RS) :
Le tableau clinique débute après l’âge de 40 ans. Il s’agit de la forme classique de PSP, caractérisée par des troubles de la marche et de l’équilibre avec chutes précoces et fréquentes, en particulier vers l’arrière, une rigidité à prédominance axiale, un rétrocolis et un syndrome frontal. Un tiers de ces patients présente des troubles de la déglutition avec des fausses routes.
Les symptômes oculaires peuvent être au début non spécifique comme une vision floue, une photophobie, une poursuite oculaire saccadée, un ralentissement des saccades verticales ou des difficultés d’accommodation (13). La PS du regard peut ne se développer que plusieurs années après. D’autres signes peuvent être associés comme des troubles de l’élocution et un blépharospasme (21, 13).
Une hyperactivité des muscles frontaux, une rétraction des paupières et un éclat du regard, contribuent à l’expression faciale de surprise des patients PSP. A l’inverse, une dystonie focale du procerus donne aux patients cette expression d’inquiétude (22). Plus de la moitié développent une modification de leur personnalité et des troubles cognitifs dans les deux premières années qui suivent le début des symptômes (23, 13). L’évolution est progressive avec une perte d’autonomie dans les trois à quatre ans qui suivent le diagnostic (20). La parole devient inintelligible, l’instabilité posturale conduit aux chutes à répétition et le patient devient dépendant du fauteuil roulant. La médiane de survie est de cinq à huit ans après le début des symptômes (21, 13). Les principales causes de décès sont les pneumopathies d’inhalation, l’embolie pulmonaire ou la détresse respiratoire d’origine centrale (24).
Dans l’étude de Respondek concernant 100 cas de PSP confirmés par autopsie, la forme RS était estimée à 50 % (25), alors que dans une étude plus récente de Takigawa, elle a été retrouvée chez 20/25 des patients, soit 80% des cas (26).
b. La paralysie supranucléaire progressive-parkinsonisme (PSP-P) : Cette forme clinique concerne environ un tiers des patients et correspond à un tableau clinique proche de celui de la MP. Chez ces patients, le syndrome parkinsonien domine le tableau clinique incluant la triade motrice et l’instabilité posturale, un début parfois asymétrique, des mouvements oculaires normaux et un certain bénéfice bien que transitoire d’un traitement par L-dopa (13). Un tremblement postural irrégulier est fréquent, parfois même associé à un tremblement de repos de 4 à 6 HZ. Bien que la rigidité axiale soit souvent un élément précoce marquant, la rigidité segmentaire des membres est plus fréquente et plus sévère que dans la PSP-SR. Les chutes et l’atteinte cognitive surviennent un peu plus tardivement. En conséquence, la durée moyenne de survie de la PSP-P excède d’environ trois ans celle de la PSP- SR (27). Le diagnostic différentiel entre une MP et une PSP-P est parfois difficile en particulier au début de la maladie. Les signes cliniques en faveur de la PSP-P sont la rapidité de l’évolution, la symptomatologie à prédominance axiale et une faible réponse à la L-dopa malgré les signes cliniques évocateurs de MPI (27).
c. PSP- Freezing :
Les critères cliniques récemment proposés pour ce syndrome comportent l’apparition précoce et progressive de troubles de la marche avec une hésitation au démarrage, puis un freezing avec enrayage cinétique de la marche, de la parole ou de l’écriture, sans tremblement, ni rigidité, ni anomalies oculomotrices, ni troubles cognitifs durant les cinq premières années d’évolution. La L-dopa n’apporte pas de bénéfice. L’imagerie cérébrale élimine une leucoencéphalopathie vasculaire de type état lacunaire ou une atteinte de la
substance blanche frontale (12). La durée moyenne d’évolution est de 11 ans. Dans son étude en 2007, William et al ont trouvé que 5,6% des patients PSP présentait une forme avec Freezing (28).
d. PSP-Syndrome cortico-basal (PSP-SCB) :
L’association d’une PSP avec un syndrome cortico-basal (PSP-SCB) est une présentation rare d’un tableau de tauopathie. Elle se caractérise par une apraxie progressive oro-buccale et des membres, asymétrique, une atteinte sensorielle corticale avec impression de membre fantôme, une dystonie d’un membre, des myoclonies et une bradykinésie non sensible à la L-dopa (26). Aucun de ces patients ne développe d’instabilité posturale ou de chutes durant la première année. Les anomalies oculomotrices sont moins sévères que dans la PSP-SR et peuvent se limiter à un ralentissement des saccades verticales. Plusieurs études ont montré que seuls 50% des patients avec un syndrome cortico-basal clinique ont une atteinte neuropathologique typique de la dégénérescence cortico-basale (DCB). Dans les études neuropathologiques, un syndrome cortico-basal a aussi été observé dans d’autres pathologies comme la maladie de Pick, la maladie d’Alzheimer et le syndrome DFT- parkinsonisme (15).
e. PSP-Frontale :
La forme frontale de la PSP comporte des troubles du comportement caractéristiques de la démence fronto-temporale (DFT) en l’absence des autres troubles moteurs de la PSP au début de l’évolution. Elle est caractérisée par une détérioration précoce et progressive de la personnalité, du comportement social et de la cognition (27). La PSP-F est une forme rare, puisque seuls trois des 66 cas de PSP autopsiés (4,5%) dans la série de la Mayo Clinic, ont présenté des changements de comportement et de la personnalité comme au cours de la DFT (28) alors que la prévalence est plus élevée dans l’étude de Respondek (29).
f. PSP-Aphasie non fluente :
La PSP-aphasie progressive non fluente (APNF) correspond cliniquement à une atteinte du langage avec un discours non fluent, des erreurs phonémiques et un agrammatisme. L’apraxie de la parole est caractéristique et secondaire à un trouble de la commande motrice. Elle est évidente dans la répétition de séries, et même si elle accompagne habituellement l’apragmatisme, elle a été rapportée comme étant un symptôme isolé et précoce d’APNF (31). Comparativement aux patients présentant d’autres tableaux d’APNF, les patients PSP-APNF ont une atteinte plus sévère de la mémoire épisodique et des praxies. L’APNF entre dans le spectre des démences fronto-temporales et peut s’associer aussi au syndrome cortico-basal (31, 32).
g. PSP- syndrome cérébelleux :
C’est une forme clinique rare de la PSP avec une ataxie cérébelleuse comme symptôme initial et cardinal avant l’apparition des signes classiques de la maladie. Dans une étude japonaise trois patients sur 22 présentaient une PSP-C, alors que dans une étude plus récente, les auteurs n’ont identifié que cinq cas de PSP-C sur 1085 cas de PSP confirmés par autopsie, dont quatre ont été diagnostiqués cliniquement comme MSA-C. Les caractéristiques de la PSP-C sont similaires à celles de la MSA-C, mais l’absence d'une dysautonomie permet de redresser le diagnostic (33).
Figure 4: Principaux signes cliniques distinctifs des différentes formes de PSP avec la maladie de parkinson (1)
E. Examens complémentaires :
a. Imagerie cérébrale : (figures 5, 6, 7)
L’imagerie cérébrale anatomique et fonctionnelle peut aider non seulement au diagnostic différentiel entre la PSP et les autres syndromes parkinsoniens, mais aussi à une meilleure connaissance des bases neurobiologiques de cette affection. Les signes IRM orientant vers une PSP est la présence d’une atrophie mésencéphalique et des pédoncules cérébelleux supérieurs. L’atrophie mésencéphalique est évoquée devant une réduction progressive du diamètre antéropostérieur du mésencéphale, avec un aplatissement progressif du toit du mésencéphale, qui prend un aspect particulier « en bec d’oiseau » appelé « signe du pingouin », « du colibri », « humminbird sign » ou « du manchot empereur », mieux visualisés en coupes sagittales médianes, fines (3 mm) T1 et T2.
Sur les coupes axiales, elle est démontrée par la présence du « signe du liseron ou ipomée » (morning glory sign) en rapport avec la concavité de la face postéro-latérale du mésencéphale. Le signe de « Mickey Mouse » correspond à un élargissement de la citerne interpédonculaire donnant un aspect de pédoncules cérébraux en oreilles de Mickey sans qu’il y ait une atrophie significative du pont.
On peut aussi noter un hypersignal T2 FLAIR et une atrophie des pédoncules cérébelleux supérieurs.
Les signes IRM évocateurs de PSP-P et de PSP-SR sont : l’atrophie du tronc cérébral avec élargissement du troisième ventricule, l’atrophie du mésencéphale, et de l’aire tegmentale ventrale, l’hypersignal de l’olive bulbaire, ainsi qu’une atrophie frontale et temporale (34).
Dans une cohorte incluant des cas confirmés de PSP, MSA, MP, DCB et des contrôles, l’IRM conventionnelle était plus spécifique mais moins sensible dans le diagnostic de PSP. Les signes « hummingbird » et « morning glory flower » avaient chacun une spécificité de 100% et une sensibilité moindre (68.4% et 50%, respectivement) (19).
Le Magnetic Resonance Parkinsonism Index (MRPI) peut être calculé, permettant ainsi de quantifier cette l’atrophie mésencéphalique : diamètre du pont/diamètre du mésencéphale × diamètre du pédoncule cérébelleux moyen/diamètre du pédoncule cérébelleux supérieur (25). Lorsque cet index est inférieur à 13,55, il est normal. Lorsqu’il est supérieur à 13,55, il y a un risque d’évolution vers une PSP. Cet index a surtout une bonne valeur discriminative entre la MP et la forme typique de PSP (SR), alors qu’il est moins discriminant dans la forme PSP-P (37). Cette atrophie mésencéphalique est significativement plus marquée dans le SR par rapport à la PSP-P et est corrélée à la sévérité de la maladie et les symptômes moteurs. L’atteinte mésencéphalique plus marquée chez les patients SR par rapport aux patients PSP-P pourrait contribuer aux différences cliniques observées entre ces deux phénotypes, en particulier les
Une cohorte incluant des cas de PSP confirmés par autopsie a révélé que le MRIP a une sensibilité de 100% et une spécificité de 99,2% à 100% pour la PSP-RS. Il est considéré comme un bon marqueur surtout au cours des stades précoces de la PSP, permettant de prédire le développement d’une forme PSP-RS chez des patients présentant un parkinsonisme et des anomalies des mouvements oculaires.(19)
Figure 5: A. Coupe axiale T1 du mésencéphale : Aspect en «oreilles de Mickey» et en «fleur de liseron» (flèches). B. Coupe sagittale du tronc cérébral : Aspect de bec d’oiseau :
« Signe du pingouin». (36)
Figure 6: signe de colibri, objectivé par l’aspect du bec d’oiseau visible sur une coupe IRM sagittale T1 (image B) (37).
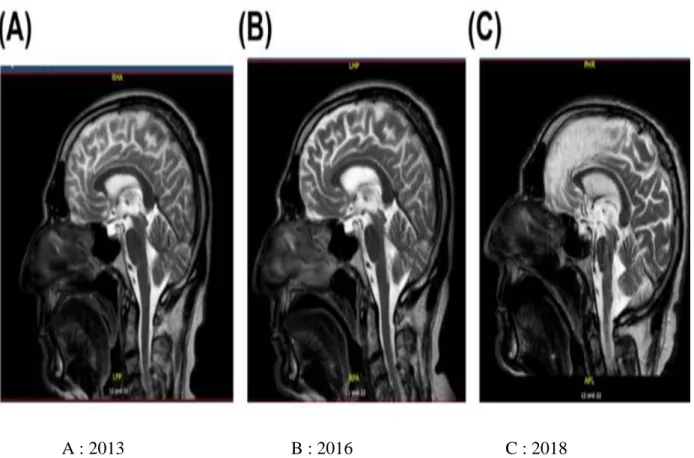
A : 2013 B : 2016 C : 2018
Figure 7: IRM cérébrale en coupes sagittales T2 d’un patient souffrant d’une PSP, objectivant une atrophie du mésencéphale (signe de colibri) et l’atrophie frontale
nettement visibles avec l’évolution de la maladie (2)
En imagerie fonctionnelle, les études du débit sanguin régional par tomographie d’émission monophotonique (TEMP) en utilisant des radioligands marqués au technétium montrent une hypoperfusion dans le cortex cingulaire antérieur et dans le cortex fronto-médian des patients PSP, en comparaison avec des sujets témoins et des patients atteints de MP. Chez les patients PSP, l’hypoperfusion s’étend sur l’aire motrice supplémentaire et le cortex préfrontal, régions impliquées dans les fonctions exécutives et les programmes moteurs. L’hypoperfusion du cortex cingulaire antérieur semble être une anomalie précoce et assez caractéristique de la PSP, tout comme l’hypoperfusion thalamique, apportant une aide au diagnostic différentiel entre la PSP et les autres syndromes parkinsoniens (38).
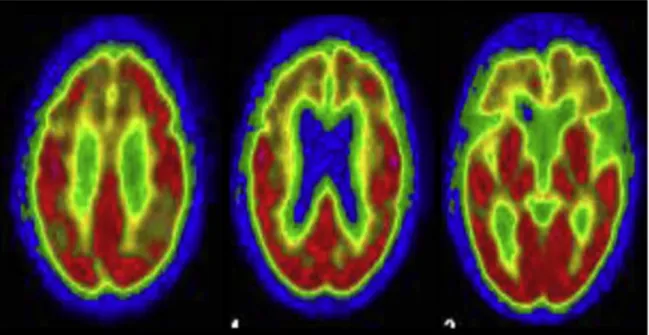
La tomographie par émission de positons au fluorodesoxyglucose (FDG-TEP) montre un hypométabolisme des régions frontales médianes et du tronc cérébral, en particulier le mésencéphale, ainsi que des aires insulaires et noyau caudé (39) (figure 8). Les anomalies en FDG-TEP varient selon le phénotype clinique, montrant un important hypométabolisme thalamique dans le SR, mais putaminal dans la PSP-P (40).
Le développement récent de radioligands spécifiques de la tauopathie en matière de TEP offre l'opportunité de faire une cartographie topographique in vivo et de quantifier l'agrégation et le dépôt de la protéine tau. Un de ces ligands est la 18F AV1451, qui se lie aux enchevêtrements et aux neurofilaments hélicoïdaux notés dans la maladie d’Alzheimer (MA). Cependant, son utilité dans la PSP est encore incertaine. Les données préliminaires ont suggéré une faible liaison du ligand à la protéine tau anormale présente sur les pièces d'autopsie de patients atteint de PSP. Il a été trouvé aussi une liaison du ligand aux zones de prédilections de la tauopathie connues dans la PSP-RS par rapport aux témoins chez les patients aux stades plus avancés de la maladie. D’autres marqueurs radiologiques à savoir le 11C PBB3, sont utilisés en matière de PSP, mais les données cliniques sont insuffisantes à l'heure actuelle pour évaluer leur utilité diagnostique (19).
Figure 8: TEP cérébrale au 18FDG chez une patiente de 65 ans atteinte de PSP-P. Notez l'hypométabolisme fronto-cingulaire mésial et orbitofrontal et la relative préservation des aires postérieures et des ganglions de la base. À noter qu'il existe ici également une
discrète atteinte pariétale supérieure bilatérale habituellement retrouvée dans les syndromes cortico-basaux (2)
b. biomarqueurs :
Au cours des dernières années, plusieurs biomarqueurs ont été évalués mais aucun n’est assez spécifique pour une détection précoce de la PSP par rapport à d’autres maladies neurodégénératives (41). Aucune n'a été réalisé dans les cas confirmés par autopsie. Plusieurs études ont évalué le taux de protéine tau dans le LCR de patients PSP, en comparaison avec les patients présentant d’autres tauopathies ou d’autres syndromes parkinsoniens neurodégénératifs comme les synucleinopathies. Le ratio 33 KDa (forme tronquée de tau) / 55 KDa (forme étendue de tau) semble être considérablement réduit dans le LCR des patients PSP par rapport à d’autres affections neurodégénératives (42). Plus récemment, il a été démontré que le poids, le type et l’ultrastructure de la protéine tau dans le
tableaux cliniques décrits ci-dessus (12). Les concentrations de phospho-tau et de tau totale dans le LCS ont été rapportés comme étant normales ou faibles par rapport aux témoins, contrairement à la MA, où ils sont élevés (19).
Plusieurs études sur le LCS ont montré une augmentation de 2 à 5 fois des concentrations de chaînes légères de neurofilaments (NfL) dans la PSP par rapport aux témoins sains, aux malades atteints de MP, démence à corps de Lewy mais pas la démence cortico basale et l’atrophie multisystématisée. Le dosage des NfL était le seul biomarqueur dans le LCS qui a changé au fil du temps lors d'un récent essai clinique multicentrique sur la PSP. Il peut désormais être mesuré dans le sang. Les patients atteints de PSP-RS ont des taux plasmatiques élevés de NfL par rapport aux témoins et patients atteints de la MP. Les concentrations plasmatiques de base de NfL preuvent prédire de la progression de la maladie au cours d'une année en corrélation avec les marqueurs cliniques et IRM. (19)
c. Exploration physiologique :
Le ralentissement des saccades verticales, plus marqué que les saccades horizontales est une caractéristique distinctive de la PSP-RS. La diminution de la vitesse des saccades est une conséquence du déficit des muscles oculaires. Des changements profonds de la vitesse des saccades ont été mis en évidence pendant une année dans deux cas de PSP confirmés par autopsie. La tomographie par cohérence optique rétinienne (OCT) peut également être un biomarqueur prometteur pour la PSP, mais elle est encore au stade des résultats préliminaires (19).
F. Critères diagnostiques :
Les critères NINDS-PSP (5) sont apparus 20 ans avant les critères de la MDS. Le diagnostic d'une PSP probable requiert la présence d’une ophtalmoplégie supranucléaire des saccades verticales et/ou une limitation du regard vers le haut associés à une instabilité posturale prédominante et des
chutes au cours de la première année de la maladie (8). Les critères associés comprennent une akinésie, une rigidité symétrique proximale et distale, une anomalie posturale du cou, en particulier un rétrocolis, une résistance à la l-dopa, une dysphagie et / ou une dysarthrie précoce et l'apparition précoce de troubles cognitivo-comportementales.
Les diagnostics « probable » et « possible » dans les critères NINDS-PSP ont une spécificité élevée (5). Cependant, le phénotype décrit dans les critères NINDS-SPS est celui d’une PSP-RS, qui ne représente qu'une forme clinique de la PSP, diminuant ainsi la sensibilité de ces critères.
Le groupe d'étude PSP de l'International Parkinson and Movement Disorder Society (MDS) a publié les critères MDS-PSP en 2017 (38) afin de déterminer les formes cliniques correspondant à la PSP ainsi que les critères utilisés en pratique courante pour le diagnostic clinique de cette pathologie.
Les critères MDS-PSP est le fruit des études et des progrès clinico-neuropathologiques concernant la compréhension de la PSP réalisés depuis la publication des critères NINDS-SPS, entrainant ainsi une augmentation de leur spécificité et sensibilité. Le groupe d'étude MDS-PSP a élaboré les nouveaux critères grâce à une revue systématique de la littérature, à la compilation de séries de cas de PSP confirmées par autopsie et à un consensus d'experts.
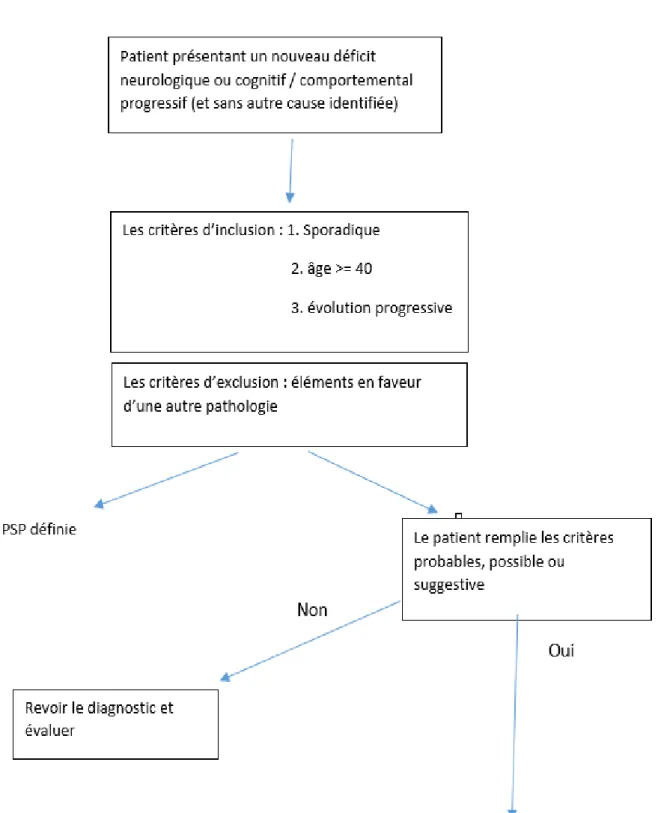
Selon les nouveaux critères de la MDS, le diagnostic clinique de PSP est évoqué chez un patient de 40 ans ou plus avec un début progressif et une progression d'une forme clinique orientant vers une PSP. Les critères d'exclusion sont divisés en critères d'exclusion obligatoires et critères d'exclusion spécifiques au contexte devant être vérifiés uniquement s'il existe des éléments suggérant un diagnostic différentiel. Les critères d'exclusion obligatoires reflètent des éléments qui suggèrent d'autres diagnostics, c'est-à-dire des troubles de la mémoire épisodiques prédominants, une dysautonomie, des
appendiculaire, un déficit des membres d’étiologie périphérique, une progression rapide, des causes identifiables d'instabilité posturale, des antécédents d'encéphalite et / ou une imagerie montrant soit une leucoencéphalopathie sévère, soit des anomalies structurelles spécifiques. Les critères d'exclusion correspondent aux données de l'imagerie, la présence de biomarqueurs orientant vers les pathologies pouvant mimer une PSP (par exemple, maladies à prion, maladie héréditaires).
L'application des critères MDS-PSP (Figure 9) nécessite une évaluation des principales caractéristiques cliniques associées à différents niveaux de certitude ou de valeur prédictive pour la PSP. Les principales caractéristiques sont classées en quatre domaines fonctionnels : troubles oculomoteurs, instabilité posturale dans les 3 ans, akinésie et dysfonctionnement cognitif (tableau 1).
Les anomalies à l’imagerie cérébrale correspondent soit à l'atrophie ou l’hypométabolisme au niveau du mésencéphale, ou la dégénérescence postsynaptique striatale-dopaminergique (5). L’application clinique des critères MDS-PSP donne un phénotype prédominant selon les symptômes retrouvés avec une évaluation du niveau de certitude (probable, possible, suggestive) (figure 9, tableaux 3, 4).
Tableau 1: Niveaux de certitude du diagnostic de PSP en fonction dessignes fonctionnels formant le noyau clinique de la maladie (38)
Tableau 3: Critères diagnostiques des différentes formes de la PSP selon les symptômes fonctionnels et anomalies IRM (38)
G. Traitement :
La prise en charge des patients atteints de PSP est souvent difficile et décevante, du fait de l’étendue des lésions rendant compte de la variabilité et la complexité des signes cliniques. Des agents actifs sur de nombreux neurostransmetteurs ont été testés, la plupart ayant montré des résultats négatifs. Les médicaments utilisés sont :
La Ldopa : Les traitements dopaminergiques sont régulièrement utilisés pour traiter les symptômes parkinsoniens. La plupart des études ont montré que si un bénéfice de la L-dopa existe en particulier dans la PSP-P, celui-ci est faible et surtout transitoire. Une revue de la littérature a évalué un taux de réponse à 26% avec une dose moyenne de l-dopa comprise entre 500 à 1000 mg par jour
Les agonistes dopaminergiques ne sont pas recommandés. Ils n’apportent pas de bénéfice, pouvant même parfois aggraver les symptômes et sont souvent mal tolérés sur le plan psychique avec la survenue d’hallucinations ou d’impulsions (40).
Les antidépresseurs tricycliques : Les doses doivent rester faibles en raison du risque de survenue d’effets indésirables comme une confusion ou des hallucinations (41).
Le zolpidem : La neurotransmission GABAergique étant sévèrement réduite dans le striatum et le pallidum des patients PSP, des agonistes GABAergiques ont été testés. Le Zolpidem administré au coucher à la dose de 5 mg a montré une amélioration des mouvements oculaires et des troubles moteurs, mais avec un bénéfice seulement transitoire (42).
La gabapentine : Elle améliore les performances des saccades, à la dose de 900mg/j sans modification des symptômes moteurs (43).
Les sérotoninergiques : Concernant la prise en charge des troubles affectifs et comportementaux, les traitements sérotoninergiques comme les inhibiteurs de la recapture de la sérotonine ou le 5-hydroxytryptophane n’ont pas fait la preuve de leur bénéfice dans la PSP. Néanmoins, ces traitements ont montré une amélioration des symptômes dépressifs et comportementaux dans d’autres tauopathies comme la démence fronto-temporale (44). Ils peuvent donc par analogie être proposés dans la PSP. Les essais cliniques avec les traitements noradrégérgiques ont montré des résultats contradictoires.
Les inhibiteurs de l’acétylcholinestérase : Une amélioration légère des troubles cognitifs a été rapportée avec les inhibiteurs de l’acétylcholinestérase. La rivastigmine a été évaluée en ouvert chez cinq patients, pendant une période de trois à six mois (45). La constation d’une légère amélioration de certaines fonctions cognitives pourrait justifier la réalisation d’études contrôlées sur un
plus grand nombre de patients. Le donépézil a été évalué dans une étude en double insu contre placebo, en cross over sur deux périodes de six semaines séparées d’un intervalle d’un mois (46). Malgré une amélioration légère des fonctions cognitives, ce traitement n’est pas recommandé chez les patients PSP du fait d’une aggravation des fonctions motrices et des activités de la vie quotidienne.
Le riluzole : Il a été évalué dans un essai clinique en double insu contre placebo, ayan inclus 362 patients PSP. Il n’a pas démontré de différence significative sur la survie à 36 mois entre le riluzole et le placebo (47).
La toxine botulinique : Elle peut être utile pour les dystonies focales cervicales (rétrocolis) et orofaciales (apraxie d’ouverture des paupières). Elle peut aussi apporter un bénéfice pour l’hypersalivation avec l’injection des glandes salivaires. Il faut cependant rester prudent du fait du risque d’aggravation des troubles de la déglutition (16).
D’autres approches sont à l’étude, ayant pour cible soit la protéine tau par inhibition de la phosphorylation ou de l’agrégation de la protéine tau, soit le dysfonctionnement de la mitochondrie par le biais du COenzyme Q10 et pyruvate (16). Le davunétide est un nouvel agent protecteur qui agirait comme stabilisateur des microtubules pouvant ralentir ainsi le processus neurodégénératif de la PSP. Des essais cliniques sont en perspective (48).
En l’absence de données scientifiques sur l’efficacité des thérapies physiques dans la PSP, leurs prescriptions sont basées sur l’expérience clinique. Les médecins rééducateurs, les kinésithérapeutes, les orthophonistes, les diététiciens et ergothérapeutes doivent être impliqués dans le traitement. Les mesures consistent en un travail de l’équilibre, la prévention des chutes et l’apprentissage du relever du sol en cas de chutes, les aides techniques à la marche, l’aménagement du domicile… La rééducation orthophonique est
boissons doit être adaptée dans le but de prévenir les fausses routes. Si besoin, une gastrostomie peut être proposée. Un suivi diététique est préconisé pour surveiller le poids et les apports nutritionnels afin de prévenir la dénutrition. Concernant les troubles oculomoteurs, des prismes peuvent être proposés au début, de même que des larmes artificielles (1).
La stimulation cérébrale profonde des noyaux sous thalamiques n’est pas indiquée dans la PSP. Quelques patients ont été implantés dans le noyau pédonculopontin avec des résultats décevants.
Ainsi, jusqu’ à maintenant, il n’y a pas de preuves suffisantes pour recommander un traitement symptomatique spécifique de la PSP. En pratique, les prescriptions doivent être adaptées individuellement à chaque patient, et une approche multidisciplinaire apparait essentielle. La prise en charge sociale et aussi indispensable pour le patient et l’aidant. De courtes hospitalisations ou séjours en institution peuvent être proposés pour un répit de l’aidant (1).
III- Matériel et méthodes :
1. Type d’étude :
Il s’agit d’une étude observationnelle, rétrospective, descriptive, menée sur une période de 5 ans (Juin 2015- Juin 2020), réalisée au service de Neurologie A et Neuropsychologie (NA) de l’hôpital des spécialités de Rabat, Centre Hospitalier Universitaire Ibn Sina.
L’étude a concerné les patients suivis à la consultation spécialisée des mouvements anormaux et de syndromes parkinsoniens du service de NA. Ces patients ont été vus directement à la consultation ou ont été hospitalisés au préalable au service de NA.
2. Critères d’inclusion et d’exclusion :
Ont été inclus tous les patients ayant un syndrome parkinsonien atypique, avec chutes, troubles oculomoteurs, et /ou avec une IRM encéphalique ayant des critères d’une PSP et répondant aux critères diagnostiques de la société des mouvements anormaux dans sa version de 2017 (MDS).
Les patients exclus de cette étude sont ceux ayant :
- un syndrome parkinsonien typique répondants aux critères diagnostiques de la maladie de Parkinson.
- un syndrome parkinsonien atypique et présentant des symptômes cliniques (troubles végétatifs, syndrome cérébelleux, démence précoce, hallucinations, apraxie...) ou radiologiques, orientant vers une atrophie multisystématisée, une démence à corps de Lewy ou une démence cortico basale.
- un syndrome parkinsonien secondaire (vasculaire, maladie de Wilson, intoxication au CO, prise de neuroleptiques…)
3. Collecte des données :
Les données cliniques ont été recueillies de manière rétrospective dans le dossier médical et analysées à l’aide d’une fiche d’exploitation préétablie (annexe 1), renseignant sur :
-Les caractéristiques démographiques : l’âge, le sexe, la profession et la couverture sociale.
-Les antécédents personnels médicaux chirurgicaux, les habitudes toxiques et l’exposition aux toxiques ainsi qu’aux antécédents familiaux.
-Les signes cliniques :
Le signe de début initial et dominant le tableau clinique, l’âge de survenu
Les troubles oculomoteurs
L’instabilité posturale
Les caractéristiques du syndrome parkinsonien
Les troubles cognitifs et du comportement.
Autres signes : troubles de la déglutition, dysarthrie spastique/hypokinétique avec hypophonie, photophobie et un syndrome pseudobulbaire.
-Les explorations paracliniques : IRM cérébrale et le bilan biologique. -Les formes cliniques
-Le traitement
-L’évolution et le suivi
Fiche d’exploitation PSP
Identité : -Nom : Prénom : -Age : -Sexe : -Profession :-Couverture sociale : CNOPS Assurance Ramed Aucune -Antécédents :
Personnels :
-Diabète : non oui - HTA : non oui
-Autres :
-habitudes toxiques : Tabagisme : non oui Alcoolisme : non oui Cannabisme : non oui -Exposition aux toxiques : pesticides non oui CO non oui - Familiaux :
MP : non oui
Alzheimer : non oui Autres : Signes cliniques : Age de début : Signes de début : Troubles oculomoteurs :
o Lenteur des saccades oculaires verticales non oui o Limitation du regard vers le haut non oui o Ophtalmoplégie supra-nucléaire non oui
Instabilité posturale :
o Chutes : précoces non oui, tardives non oui Akinésie :
o Marche à petite pas : non oui
o Tremblement : non oui . Localisation : MS Dt Gche MI Dt Gche o Bradykinésie des membres non oui. Symétrique asymétrique o Rigidité axiale et des membres non oui
o Freezing progressif de la marche non oui, précoce tardif Troubles cognitifs :
-Troubles de la parole : non oui o Apraxie de la parole non oui
o Diminution de la fluence verbale agrammatisme et erreurs phonémiques non oui
-Syndrome frontal et ou trouble du comportement :
o Apathie :
o Bradyfrénie :
o Syndrome dysexécutif :
o Diminution de la fluence verbale :
o Impulsivité, désinhibition, persévération :
-Syndrome cortico basal :
o Apraxie orobucale ou d’un membre
o Déficit sensoriel
o Main étrangère
o Rigidité, akinésie ou myoclonies d’un membre
Autres :
o Troubles de la déglutition : non oui
o Dysarthrie spastique, hypokinétique avec hypophonie : non oui o Photophobie : oui non
Paracliniques : IRM cérébrale :
o L’atrophie corticale oui non o L’atrophie mésencéphalique oui non
o L’hypersignal en T2 du pallidum interne oui non o Discret hypersignal en T2 bordant le putamen. Oui non o L’élargissement de l’aqueduc de Sylvius oui non o L’élargissement du troisième Ventricule oui non
o Hypersignal punctiforme en T2 de la partie haute du mésencéphale oui non
Biologie
o Pl déplétive : amélioration oui non o NFS :
o Glycémie à jeun : diabète non oui
o TSH : normal hypothyroïdie hyperthyroïdie o Fonction rénale : normale IRC IRA
o Vit B12 : normal carence NF o Vit B9
o Sérologies virales : VHB : VHC : VIH : o VS : normale accélérée
Radio poumon : Autres :
Formes cliniques :
o PSP- RS (IP+ chutes précoces+ paralysie de la verticalité) o PSP-IP
o PSP de type parkinsonien (PSP-P)
o PSP-F
o PSP-akinésie pure avec « freezing » de la marche o PSP-syndrome cortico-basal
o PSP-aphasie progressive non fluente ou dysarthrie apraxique
o PSP-SLP o PSP –syndrome cérébelleux Traitement : Résistance à L- Dopa Autres : Evolution et suivie :
IV- Résultats :
A/ Observations cliniques :
Douze cas de PSP ont été recensés. Les observations cliniques des patients sont les suivantes :
1. Mr B. S :
Il s’agit d’un patient de 41 ans, lettré, ayant comme antécédents une hypertension artérielle (HTA) sous Amlodipine, une dyslipidémie depuis trois ans, un accident vasculaire cérébral ischémique en 2014, un tabagisme chronique, un cannabisme et éthylisme sevrés depuis sept ans, hospitalisé au service de neurologie A en 2017 pour mise au point diagnostic de troubles neurologiques.
Le début de sa symptomatologie remonte à 2016, par l’apparition de troubles de la posture et de la marche, des troubles d’attention et une incontinence urinaire. L’examen clinique objectivait une akinésie, une rigidité extra pyramidale aux quatre membres et une limitation des mouvements oculaires vers le haut. Par ailleurs, le malade présentait des épisodes de pleurs spasmodiques, avec un test MMSE coté à 20/30. L’IRMC a noté une dilatation ventriculaire et une leuco-encéphalopathie hypertensive minime. Le bilan biologique était normal, la ponction lombaire déplétive était sans effet, ainsi que la L-Dopa instaurée à dose progressive. Le diagnostic de paralysie supra nucléaire progressive forme Richardson (PSP-RS) a été retenu. Le malade a été perdu de vue.
2. Mme F. B :
Il s’agit d’une patiente de 55 ans, sans antécédents notables, qui présente depuis 2018, des chutes fréquentes, des troubles de l’humeur avec un retrait social et des troubles oculomoteurs à type d’une paralysie de la verticalité du regard vers le haut et une paralysie de la convergence. La 1ère consultation date
de février 2020. L’examen clinique a objectivé une bradykinésie, une rigidité extra pyramidale latéralisé à droite, une paralysie de la verticalité du regard vers le haut, avec à l’examen des fonctions supérieurs un ralentissement psychomoteur, des troubles attentionnels et une humeur dépressive. Le bilan biologique ainsi que L’IRM C étaient normaux. Le diagnostic de PSP-RS a été retenu. La patiente a été mise sous Madopar à raison d’un quart de comprimé par jour puis perdue de vue.
3. Mme B. F :
Il s’agit d’une patiente de 70 ans, suivie pour une ostéoporose et une HTA sous traitement, ayant un oncle suivi pour une maladie de Parkinson. Le début de sa symptomatologie remonte à 2010 par l’apparition de troubles de la parole et un ralentissement moteur, aggravé en 2016 par des troubles de la posture avec rétropulsion sans chutes. La 1ère consultation date de Juin 2016. L’examen clinique lors sa 1er consultation a objectivé une akinésie modérée, une diminution du ballant à droite une rigidité du membre supérieurs droit modérée, une paralysie de la verticalité vers le haut et une dysarthrie extrapyramidale. Le test de la poussé était positif cependant la patiente arrivait à rétablir son équilibre. l’IRM C était normal. La patiente a été mise progressivement sous L-Dopa jusqu’à une dose de 800mg/j associé au Trivastal 150 mg/j avec une réponse modérée à la dopathérapie. Le diagnostic de PSP- RS probable a été retenu. La patiente n’a jamais été hospitalisée au service. Elle a été perdue de vue.
4. Mr C. E :
Il s’agit d’un patient de 70 ans, ayant comme antécédents un lymphome type T traité par chimiothérapie en 2016 et un cancer de la langue traité par radiothérapie en 1998 avec présence comme séquelles d’une hypoacousie et d’une diplopie.
Le début de la symptomatologie remonte à Juillet 2018, par l’apparition de troubles de la marche à type de freezing d’aggravation progressive et occasionnant des chutes fréquentes. Des oublis à mesure et des troubles urinaires sont apparus en 2018. La 1ère hospitalisation date de Septembre 2018. L’examen clinique a trouvé une bradykinésie latéralisée à gauche, une hypertonie extrapyramidale de l’hémicorps gauche et une ophtalmologie avec un limitation du regard vers le haut, avec des troubles attentionnels à l’examen de fonctions supérieures. Le bilan biologique était normal. L’IRM C a objectivé une dilatation ventriculaire sans atrophie corticale avec une leucoencéphalopathie hypertensive minime. Le patient fut mis sous LDopa, puis il a été perdu de vue. Une forme PSP- freezing a été retenue.
5. Mr E. B :
Il s’agit d’un patient de 61 ans, ayant comme antécédents un tabagisme, cannabisme et alcoolisme sevré il y a 4ans. Le début de la symptomatologie remonte à 2014 par l'apparition de trouble de la posture avec des chutes fréquentes, des oublis à mesure et une labilité de l’humeur. La 1ère consultation date de Juin 2016. L’examen clinique a trouvé une akinésie globale, une hypertonie extra pyramidale prédominant aux membres supérieurs et en axial, une dysarthrie extra pyramidale et des troubles de l’humeur d’ordre dépressifs. L’IRM C a objectivé une atrophie corticale avec un élargissement du système ventriculaire. Le bilan biologique ainsi que la ponction lombaire étaient normaux. Le patient fut mis sous LDopa, puis a été perdu de vue depuis sa sortie de l’hôpital. La forme PSP- RS a été retenue.
6. Mr E. A :
Il s’agit d’un patient de 61 ans, diabétique type 2 sous antidiabétiques oraux, suivi pour un goitre multi nodulaire en euthyroïdie et tabagique et d’éthylique chronique sevré depuis 10 ans. Le début de la symptomatologie remonte à 2015, par l’apparition de chutes fréquentes, associé à des troubles de
la parole, des rires et des pleurs spasmodiques, ainsi que des troubles de la déglutition et des oublis à mesure. La 1ère hospitalisation au service date de Juin 2016. L’examen clinique a objectivé une bradykinésie sans tremblement, une hypertonie axiale, une limitation du regard vers le haut, une dysarthrie extrapyramidal sévère et une humeur dépressive. L’IRM encéphalique a objectivé une atrophie mésencéphalique avec un aspect de « bec d‘oiseau ». Le bilan biologique était normal. La forme PSP-RS fut retenue. Le patient a été mis sous LDopa à la dose de 800mg/j. l’évolution a été marquée par une aggravation des troubles de la parole, le patient est complétement anarthrique. Les troubles oculomoteurs se sont également aggravés avec une paralysie totale de la verticalité et une fixité du regard. Le patient présente aussi des fausses routes aux liquides et aux solides. Une gastrostomie d’alimentation a été proposée mais fut refusée par le patient. Le patient est toujours suivi à la consultation.
7. Mr E. M :
Il s’agit d’un patient de 68 ans sans antécédents pathologiques notables. Le début de la symptomatologie remonte à 2016, par l’apparition de troubles de la parole, d’une dysphagie, d’un syndrome peudobulbaire, compliqué de troubles de la marche avec des chutes fréquentes. La 1ère hospitalisation date de Mai 2019. L’examen clinque a objectivé une bradykinésie et une rigidité des quatre membres modérés, une rigidité axiale (attitude en hyperextension de la tête et du cou), une ophtalmoplégie à type de limitation du regard vers le haut et une dysarthrie d’allure paralytique. L’IRM encéphalique a objectivé une atrophie mésencéphalique objectivée par l’atrophie du sillon inter pédonculaire. Le patient fut mis sous LDopa à la dose de 800mg/j sans amélioration. La forme PSP-RS fut retenue. L’évolution a été marquée par l’aggravation de la bradykinésie qui est devenue sévère ainsi que les chutes qui sont devenues très fréquentes. La paralysie de la verticalité du regard est devenue totales associée à une limitation de la latéralité vers la gauche. La dysarthrie est devenue très sévère avec une parole quasi incompréhensible. La gastrostomie d’alimentation fut refusée par le patient.