Association Between Tumor Necrosis Factor Receptor II and
Familial, but Not Sporadic, Rheumatoid Arthritis
Evidence for Genetic Heterogeneity
Philippe Dieude´,
1Elisabeth Petit,
1Se´verine Cailleau-Moindrault,
1Jose´ Osorio,
1Ce´line Pierlot,
1Maria Martinez,
2Sabine Faure´,
3Olivier Alibert,
4Sandra Lasbleiz,
5Claudia de Toma,
6Thomas Bardin,
7Bernard Prum,
8and Franc¸ois Corne´lis,
7for the European Consortium on
Rheumatoid Arthritis Families
Objective. Tumor necrosis factor␣ (TNF␣) binds the receptors TNFRI and TNFRII. Results of genome scans have suggested that TNFR2 is a candidate rheu-matoid arthritis (RA) locus. A case–control study in a UK Caucasian population has shown an association
between a TNFR2 genotype (196R/R in exon 6) and familial, but not sporadic, RA. The present study was undertaken to test this association in the French Cau-casian population.
Methods. To test for an association in sporadic
RA, 100 families were genotyped for the 196M/R poly-morphism and analyzed using the transmission disequili-brium test and haplotype relative risk. To test for an association in familial RA, RA index cases from 100 affected sibpair (ASP) families were genotyped for 196M/R. Linkage analysis was performed with 3 TNFR2 microsatellite markers.
Results. The TNFR2 196R/R genotype was not
associated with sporadic RA (odds ratio [OR] 0.59, Pⴝ 0.72), but was associated with familial RA (OR 4.0, Pⴝ 0.026). The association was most marked in the context of TNFR2 “twin-like” RA sibs (affected sibs sharing both TNFR2 haplotypes) (OR 9.2, Pⴝ 0.0017). Linkage analysis results were consistent with the association; most of the TNFR2 linkage evidence was found in the subgroup of families with 196R/R ASP index cases.
Conclusion. This study is the first to replicate
evidence of the involvement of TNFR2 in RA genetic heterogeneity. Our data refine the initial hypothesis, to suggest that a TNFR2 recessive factor, in linkage disequi-librium with the 196R allele, plays a major role in a subset of families with multiple cases of RA.
Genetic and environmental factors are thought to be involved in the pathogenesis of rheumatoid arthritis (RA). The prevalence of the disease is increased in first-degree relatives of RA patients, with an estimated sibling risk between 1.8% and 12.1% (for review, see ref.
Supported in part by the Fondation pour la Recherche Me´dicale, Association Franc¸aise des Polyarthritiques, Association de Recherche pour la Polyarthrite, Socie´te´ Franc¸aise de Rhumatologie, Association Rhumatisme et Travail, Genopole, Conseil Re´gional Ile de France, Ministe`re de la Recherche et de l’Enseignement Supe´rieur, Schering-Plough, Pharmacia, Pfizer, and Wyeth. Dr. Dieude´’s work was supported by a grant from the Acade´mie Nationale de Me´decine. ECRAF, the European Consortium on Rheumatoid Arthritis Families, was initiated, with funding from the European Commission (BIOMED2), by T. Bardin, D. Charron, F. Corne´lis (coordinator), S. Faure´, D. Kuntz, M. Martinez, J. F. Prudhomme, and J. Weissenbach (France); R. Westhovens and J. Dequeker (Belgium); A. Balsa and D. Pascuale-Salcedo (Spain); M. Spyropoulou and C. Stavropoulos (Greece); P. Migliorini and S. Bombardieri (Italy); P. Barrera and L. van de Putte (The Netherlands); and H. Alves and A. Lopes-Vaz (Portugal).
1Philippe Dieude´, MD, Elisabeth Petit, PhD, Se´verine
Cailleau-Moindrault, Jose´ Osorio, Ce´line Pierlot: GenHotel, Evry-Genopole, France;2Maria Martinez, PhD: INSERM U358, Hoˆpital
Saint-Louis, Paris, France; 3Sabine Faure´, PhD: Ge´ne´thon, Evry,
France; 4Olivier Alibert: Ge´ne´thon, Evry, France, and CEA,
Evry-Genopole, France; 5Sandra Lasbleiz, MD: Centre Viggo-Petersen,
Hoˆpital Lariboisie`re, Assistance Publique des Hoˆpitaux de Paris, Paris, France; 6Claudia de Toma, PhD: Fondation Jean Dausset CEPH,
Paris, France;7Thomas Bardin, MD, Franc¸ois Corne´lis, MD, PhD:
GenHotel, Evry-Genopole, France, and Centre Viggo-Petersen, Hoˆ-pital Lariboisie`re, Assistance Publique des Hoˆpitaux de Paris, Paris, France; 8Bernard Prum, PhD: Laboratoire Statistique et Ge´noˆme,
Evry-Genopole, France.
Address correspondence and reprint requests to Philippe Dieude´, MD, GenHotel Laboratoire de Recherche Europe´en pour la Polyarthrite Rhumatoı¨de, Evry-Genopole, ECRAF-Universite´ Paris 7-Universite´ Evry, 2 rue Gaston Cre´mieux, CP 5727 91057, Evry, France. E-mail: dieude@polyarthrite.net.
Submitted for publication June 22, 2001; accepted in revised form October 9, 2001.
1), providing a prevalence ratio versus that in the general population of 3–15 (2,3). Twin studies provide evidence for genetic factors in RA, with a concordance rate in monozygotic twins 3.5–5.2 times higher than that in same-sex dizygotic twins (4–6). The first known RA gene was HLA–DRB1, coding for the protein HLA– DR1 (7). The homology between HLA–DRB1 suscep-tibility alleles led to the “shared epitope” hypothesis (8), but the pathophysiologic mechanism remains unknown. The HLA locus accounts for one-third of the genetic component of RA (1,9).
The search for non–HLA-linked susceptibility genes has been hampered by the clinical and genetic heterogeneity of the disease. Multiple HLA–DRB1 al-leles are associated with RA in different populations, and as many as one-fourth of RA patients do not have any of these alleles. It is likely that genetic heterogeneity will also be found for non-HLA RA genetic factors, so the RA phenotype could be related to several combina-tions of susceptibility alleles.
Linkage analysis of affected sibpairs (ASPs) is useful to define loci with suggestive linkage, with an increased probability of harboring RA genes. Recently, genome-wide linkage studies of RA sibpairs indicated a candidate region at 1p36 (10,11), which includes the tumor necrosis factor receptor (TNFR) p75 gene (TNFR2) (12). Results of further studies strengthened the linkage evidence in European (13), but not in North American (14) populations.
TNF␣, which is found at elevated levels in RA serum and synovium (15,16), contributes to the chronic inflammatory response. Notably, therapy directed against TNF␣ has been found to be efficacious in RA (17,18). TNF␣ acts via binding to cell surface receptors TNFRI (TNFR p55) and TNFRII (TNFR p75). TNFRII belongs to the TNFR superfamily characterized by cysteine-rich extracellular domains (for review, see ref. 19). In RA synovium, but not in osteoarthritis (OA) synovium, TNFRII is strongly expressed at the cartilage– pannus junction (20). The protein is also produced naturally as a soluble form (sTNFRII). Patients with RA have circulating levels of sTNFRII higher than those observed in patients with OA or non-RA inflammatory arthritis (21–23). The soluble receptor inhibits TNF␣ action by competing with cell surface receptors in bind-ing TNF␣, thereby blocking its biologic effects. Indeed, RA patients benefit from infusion of TNFRII (17). Based on these findings, the TNFR2 gene is considered to be a strong candidate as an RA susceptibility gene, with the hypothesis that its functional characteristics are allele dependent.
Santee and Owen-Schaub (24) characterized the complete structure of the human TNFR2 gene, which spans nearly 43 kb. The gene has 10 exons and 9 introns. A biallelic polymorphism in exon 6 has been described previously (25). This single-nucleotide polymorphism (ATG 3 AGG) resulted in a nonconservative amino acid substitution (methionine [M] 3 arginine [R] at codon 196 [196M/R]). Exon 6 encodes a small portion of the transmembrane region and the proteolytic cleavage site that enables production of sTNFR2 (26). Receptor shedding provides a mechanism for down-regulation of the receptor at the cell surface and a means of releasing the physiologically active soluble receptor, which acts as a receptor antagonist. Increased shedding explains the high circulating TNFRII levels in the serum and synovial fluid of patients with active RA (21–23). However, it is not known whether shedding is influenced by the exon-6 polymorphism.
Two RA case–control association studies regard-ing this polymorphism were recently reported (27,28). The first, with a large Japanese population, revealed no significant association (27). In the second study, with a Caucasian UK population, no association was observed for sporadic RA, but an association between the 196R/R genotype and familial RA (RA with affected sibs) was described (odds ratio [OR]) 2.96, 95% confidence inter-val 1.3–6.6, P ⫽ 0.007) (28). These findings led to the hypothesis that the TNFRII 196R/R genotype might contribute to RA genetic susceptibility in a subgroup of patients with a family history of RA. The aim of the present study was to provide an independent test of this hypothesis based on RA families in the French Cauca-sian population.
PATIENTS AND METHODS
Study design. A family-based association study was performed to investigate the association in sporadic RA. To test for association in familial RA, RA index cases from ASP families previously genotyped for the TNFR2 locus were studied, taking into account the linkage analysis of those families.
Patients.All subjects provided informed consent, and the ethics committee of Hoˆpital Biceˆtre (Kremlin-Biceˆtre, France) approved the study. RA patients and controls were recruited through a national media campaign followed by selection of individuals who fulfilled the American College of Rheumatology (ACR; formerly, the American Rheumatism Association) 1987 criteria for RA (29) according to their physicians. All clinical data were reviewed by rheumatologists from Centre Viggo-Petersen, Hoˆpital Lariboisie`re, Assistance Publique des Hoˆpitaux de Paris.
Sporadic RA sample. One hundred French Caucasian
living RA patient and both living parents, with no other familial history of RA, were studied. All patients fulfilled the ACR 1987 criteria for RA. Among the RA patients tested, 87 were women and 13 were men. The mean age at disease onset was 29.2 years. Sixty-eight percent of the patients were rheu-matoid factor (RF) positive, 74.5% had 1 or 2 HLA–DRB1 susceptibility alleles (“shared epitope”), and 94% presented with erosive disease.
Familial RA sample. The study group from ASP
fami-lies consisted of 100 unrelated French Caucasians (the 4 grandparents were European Caucasian). Among the ASP index patients tested (all of whom fulfilled the ACR 1987 criteria for RA), 77 were women and 23 were men. The mean age at disease onset was 39.8 years. Eighty-five percent of the patients were RF positive, 76% had 1 or 2 HLA–DRB1 susceptibility alleles (“shared epitope”), and 86% presented with erosive disease. These ASP families had been genotyped for microsatellite markers in the TNFR2 locus: 1 intragenic microsatellite marker located within intron 4 (26) and 2 flanking microsatellite markers (D1S2667 and D1S228) spaced at 5.3 cM and 0.2 cM on either side of the TNFR2 locus, with heterozygosity of 82% and 76%, respectively (13,30).
Power calculation. A sample of 100 patients with familial RA and 100 controls provides a power of 50% to detect the association between familial RA and the 196 R/R genotype that was described in the UK population (28) with an OR of 2.96 at the 5% significance level, assuming a 196R/R genotype frequency in controls of 5%.
Molecular biology methods.Genomic DNA used for genotyping was purified from fresh peripheral blood leuko-cytes by standard methods. Genotyping of TNFR2 196 morphism was performed by restriction fragment length poly-morphism (Nla III), as previously described (31). The substitution at codon 196, i.e., ATG (methionine) 3 AGG (arginine), eliminates the Nla III restriction site. Each geno-type was interpreted independently by 2 individuals (PD and EP).
Analysis methods.The hypothesis tested was that there is an association between the 196R/R genotype and RA, confined to the context of familial RA. In all association, linkage, and equilibrium analyses, the chi-square test with 1 degree of freedom was used. P values less than 0.05 were considered significant.
Test for association in sporadic RA. The sample of
patients with sporadic RA and their families was used to test for an association in the absence of familial RA. Association analyses were performed using the transmission disequilibrium test (TDT) and haplotype relative risk (HRR) (32,33). The TDT tested the excess in transmission of the 196R allele from heterozygous parents to affected offspring, compared with Mendel’s expectation (50%) (32). A subanalysis was per-formed for 2 subgroups of parents with defined genotype combinations (196R/R⫻ 196 R/M, and 196R/M ⫻ 196R/M) that could lead to the 196R/R genotype offspring. As in a case–control study, HRR tested for an excess of the 196R/R genotype in RA cases compared with the “controls” (recon-structed genotype from nontransmitted parental alleles).
Test for association in familial RA. The 196R/R
geno-type frequencies in RA index cases from ASP families were compared with those in both RA patients from sporadic RA families and controls derived from sporadic RA families. To
explore the hypothesis of involvement of the 196R/R genotype specifically in familial RA, i.e., not an association with the 196R/R genotype per se, we defined a subgroup of ASP index cases who were more likely to have a putative factor at the
TNFR2 locus. This subgroup consisted of RA index cases with
a “twin-like” RA sib: a TNFR2 genetically identical affected sib (RA cases for whom the affected sib had an identical genotype at the 3 TNFR2 microsatellites). The 196R/R genotype fre-quencies were compared between the subgroup of RA index cases with a “twin-like” sib and the remaining subgroup.
Test for linkage in familial RA. In the hypothesis of
involvement of the 196R/R genotype in familial RA, linkage is expected at the TNFR2 locus in ASP families in which the index RA case has the 196R/R genotype. Linkage analysis was performed for the TNFR2 intragenic microsatellite and both flanking markers in all families, in families in which the index case had the 196R/R genotype, and in the remaining families. ASP analysis was conducted with the ANALYZE package (from J. Terwilliger, Columbia University, New York, NY) using the program SIBPALNA 1.1; P values less than 0.05 were considered suggestive. In comparisons of ASP sharing between the 2 subgroups, P values less than 0.05 were considered significant.
Linkage disequilibrium. Allelic association between
196R and the alleles of the 3 TNFR2 microsatellite markers was investigated, comparing the frequency of those alleles between the individuals with 196R/R and the individuals with 196M/M. Linkage disequilibrium between microsatellite mark-ers was investigated similarly.
Hardy-Weinberg equilibrium. The Hardy-Weinberg
equilibrium of the TNFRII 196R/M polymorphism was inves-tigated for all analyzed populations.
RESULTS
Results of test for association in sporadic RA.The allele frequencies of 196M and 196R alleles among the 100 cases of sporadic RA were 79% and 21%, respectively. The transmission of the 196R allele from heterozygous parents (33 of 74 times [44.6%]) showed no excess over what would be expected by Mendel’s law (50%) (Table 1). The subana-lysis performed on the subgroup of families for which the parents could have 196R/R offspring revealed no excess of
Table 1. Absence of association of the TNFR2 196R allele with susceptibility to sporadic rheumatoid arthritis (RA) in the French Caucasian population: transmission of TNFR2 196R allele to RA offspring
Transmitted,
no. (%) Not transmitted,no. (%) P
All heterozygous parents
(n⫽ 74) 33 (45) 41 (55) 0.35
Parents 196R/M⫻ 196R/M
(n⫽ 25) 12 (48) 13 (52) 0.68
Parents 196R/R⫻ 196R/M
196R transmission (Table 1). HRR analysis of the 196R/R genotype revealed the presence of the 196R/R genotype in 3 RA cases (3%) and 5 control genotypes (5%) (OR 0.59, P⫽ 0.72).
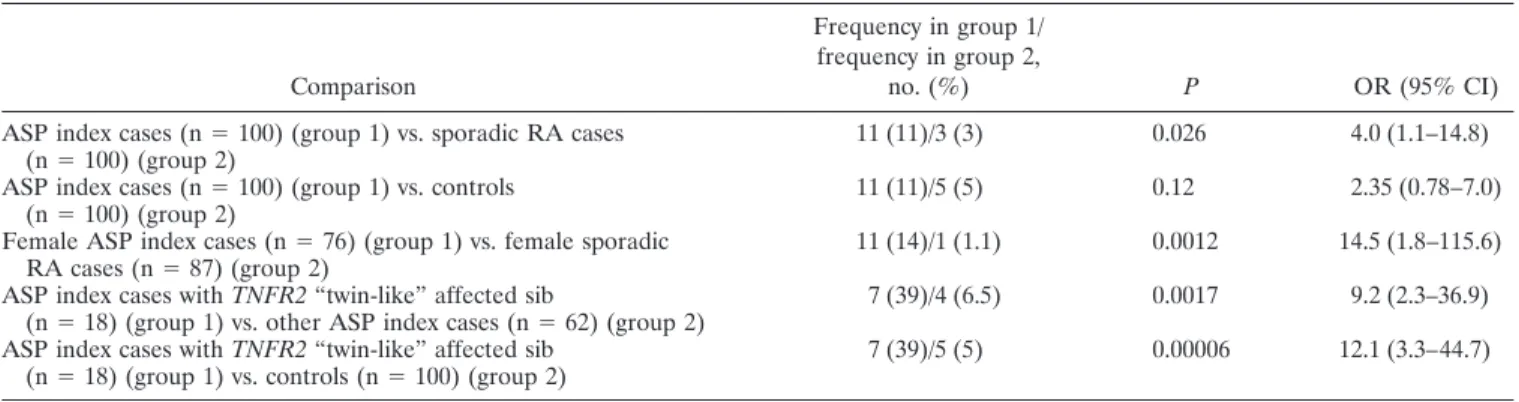
Results of test for association in familial RA.The frequencies of the 196M and 196R alleles in RA index cases from ASP families were 72% and 28%, respec-tively, similar to the frequencies among sporadic RA cases. The 196R/R genotype was significantly more frequent in RA ASP index cases than in sporadic RA cases (11% versus 3%; P ⫽ 0.026). This genotype was also more frequent in RA ASP index cases than in controls (11% versus 5%; P ⫽ 0.12) (Table 2). This difference was not related to the difference in the sex distribution among the familial and sporadic RA groups: results were similar in the subgroup of female subjects (14% versus 1.1%; P⫽ 0.0012) (Table 2).
To further test the hypothesis that the 196R/R genotype is associated with familial RA, we compared 196R/R genotype frequencies between the TNFR2 “twin-like” subgroup of RA ASP index cases, which had a higher probability of harboring TNFR2 RA factors, and the remaining RA ASP index cases. Of 80 RA index cases from ASP families with complete genotyping data for the 3 TNFR2 markers, there were 18 RA cases with “twin-like” TNFR2 RA sibs. Seven of those had the 196R/R genotype, compared with 4 of the 62 remaining RA cases (39% versus 6.5%; P ⫽ 0.0017) (Table 2), indicating a major increase in the frequency of the suspected genotype in the “twin-like” RA sample. Com-parison of the 196R/R genotype frequencies between this sample and the controls provided further evidence of a strong association with RA in this RA subgroup (39% versus 5%; P⫽ 0.00006) (Table 2). No clinical or HLA–DRB1 differences were found between the sub-group of 7 ASP index cases who had “twin-like” affected
sibs and had 196R/R genotype, versus the other RA cases who had no RA sib sharing both TNFR2 haplo-types and did not have the 196R/R genotype (data not shown).
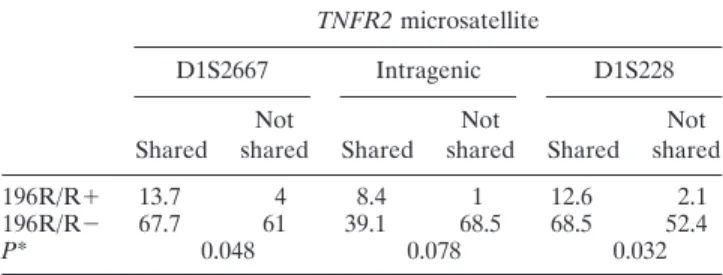
Results of test for linkage in familial RA.Results of ASP analysis suggested linkage at the TNFR2 locus (Table 3). Most of the evidence came from the subgroup of families with 196R/R index cases. Comparison of allele sharing between the 196R/R and non-196R/R subgroups showed a significant difference (Table 4), consistent with the hypothesis of genetic heterogeneity. Results of linkage disequilibrium and Hardy-Weinberg equilibrium tests. No linkage disequilibrium was found between the 196R allele and any allele of the 3 microsatellites tested. No linkage disequilibrium was found between the microsatellite alleles, providing a combined heterozygosity of 97% at the TNFR2 locus. The genotype distributions in the RA ASP index cases, the sporadic RA cases, and the “reconstructed controls” were compatible with Hardy-Weinberg equilibrium.
DISCUSSION
The aim of this study was to test, in the French Caucasian population, the hypothesis of involvement of
Table 2. Association of the TNFR2 196R/R genotype with susceptibility to familial RA in the French Caucasian population*
Comparison
Frequency in group 1/ frequency in group 2,
no. (%) P OR (95% CI)
ASP index cases (n⫽ 100) (group 1) vs. sporadic RA cases
(n⫽ 100) (group 2) 11 (11)/3 (3) 0.026 4.0 (1.1–14.8)
ASP index cases (n⫽ 100) (group 1) vs. controls
(n⫽ 100) (group 2) 11 (11)/5 (5) 0.12 2.35 (0.78–7.0)
Female ASP index cases (n⫽ 76) (group 1) vs. female sporadic
RA cases (n⫽ 87) (group 2) 11 (14)/1 (1.1) 0.0012 14.5 (1.8–115.6)
ASP index cases with TNFR2 “twin-like” affected sib
(n⫽ 18) (group 1) vs. other ASP index cases (n ⫽ 62) (group 2) 7 (39)/4 (6.5) 0.0017 9.2 (2.3–36.9) ASP index cases with TNFR2 “twin-like” affected sib
(n⫽ 18) (group 1) vs. controls (n ⫽ 100) (group 2) 7 (39)/5 (5) 0.00006 12.1 (3.3–44.7)
* Sporadic rheumatoid arthritis (RA) refers to cases in which other family members are not affected. TNFR2 “twin-like” affected sibs are affected sibs who are genetically identical to the index case at the 3 TNFR2 microsatellites. OR⫽ odds ratio; 95% CI ⫽ 95% confidence interval; ASP ⫽ affected sibpair.
Table 3. Significance (P values) of the results of linkage analysis at the TNFR2 locus in families studied*
TNFR2 microsatellite
D1S2667 Intragenic D1S228
All families 0.08 0.05 0.006
Families in which ASP index case
had the 196R/R genotype 0.008 0.002 0.001
Families in which ASP index case did not have the 196R/R genotype
0.28 0.21 0.05
TNFR2 in RA genetic heterogeneity. Our findings sup-port and refine the initial observation made by Barton et al in the UK population (28). We observed an associa-tion between the TNFR2 196R/R genotype and familial RA only, with no association in sporadic disease (11% of index cases in RA multiplex families, 3% of sporadic RA cases, and 5% of controls). We have therefore replicated the initial observation in a second European population. Additional support was provided by the results of link-age analysis, which were consistent with the association. The restriction of the association to the context of familial disease suggests that this genotype is not the susceptibility factor, but a marker for it. In accordance with this, the association was most pronounced in the context of TNFR2 “twin-like” RA sibpairs, as if a TNFR2 genetic factor was concentrated in those families (39% of “twin-like” cases, versus 6.5% of remaining cases).
Thus, the data are consistent with the presence of a recessive TNFR2 factor, in linkage disequilibrium with the 196R allele, that is involved in RA genetic hetero-geneity and plays a major role in a subset of familial RA. Since no obvious correlation with clinical features or with the HLA–DRB1 genotype was seen, this putative factor would be independent from HLA–DRB1, and lead to a phenotype that is clinically indistinguishable from sporadic RA. The recent discovery of a new genetic factor for Crohn’s disease, identified through linkage disequilibrium of rare disease alleles with common polymorphisms in a subset of families, using DNA from index cases with “twin-like” affected sibs (34), strength-ens the hypothesis. Alternatively, since not only genetic factors but also environmental factors are accumulated in multiplex families, it could be hypothetized that family-specific environmental factors interact with the TNFR2 196R allele, the TNFR2 196R/R genotype being itself the RA factor.
Our data are consistent with results of previous studies in other populations, with association observed not in sporadic RA (27,28) but only in familial RA, in a
similar family context and with similar genotype fre-quencies (14% in RA versus 7% in controls in the UK population) (28). The small number of families studied could explain the reported absence of suggestion of linkage in other general analyses of Caucasian samples from the North American Rheumatoid Arthritis Con-sortium (NARAC) and the UK (14,35). The suggestion of linkage at the TNFR2 locus in the Japanese study and the absence of association in Japanese sporadic RA cases (27) are compatible with a similar involvement of TNFR2 in the Japanese population.
This study represents the first replication of the initial finding in another Caucasian population. Other replication studies in other European populations are warranted before it can be concluded that TNFR2 is a factor involved in RA genetic heterogeneity in Cauca-sians. We were fortunate to make this observation with an estimated power of only 50%, a large sample size being required to reach appropriate power. It would be worthwhile to investigate the familial ASP samples from the UK, the NARAC, and Japan to confirm a major role of this putative TNFR2 factor in a significant subset of TNFR2 “twin-like” RA sibpairs.
To identify the putative TNFR2 factor involved in RA genetic heterogeneity, investigation could focus on the small number of families with evidence of TNFR2 linkage, searching for a mutation in linkage disequili-brium with the 196R allele. The absence of linkage disequilibrium seen in this study suggests that any region of strong linkage disequilibrium around 196R is small, facilitating the search. This TNFR2 factor, although involved in only a small number of RA patients, might be of major pathophysiologic interest, especially in view of the therapeutic effect of anti-TNF drugs. It would be particularly interesting to investigate the relationship between this TNFR2 polymorphism and treatment re-sponse.
This factor could help identify new interacting genetic factors. To aid in such efforts, the genotype data described here are publicly available through the Gen-Hotel program (www.polyarthrite.net). Moreover, the strategy used in this study, taking advantage of a familial sample previously genotyped for markers closely linked to the candidate gene, could help solve the puzzle of RA genetics. Implementation of this strategy is possible for other investigators, through the GenHotel program (www.polyarthrite.net).
In conclusion, our findings support and refine the initial observation of TNFR2 as a newly identified RA genetic factor. Further investigations using the strategy described here could help unravel the genetic hetero-geneity of RA.
Table 4. Comparison between the 196R/R⫹ and 196R/R⫺ affected sibpair subgroups in terms of TNFR2-microsatellite allele sharing
TNFR2 microsatellite
D1S2667 Intragenic D1S228
Shared sharedNot Shared sharedNot Shared sharedNot
196R/R⫹ 13.7 4 8.4 1 12.6 2.1
196R/R⫺ 67.7 61 39.1 68.5 68.5 52.4
P* 0.048 0.078 0.032
* Significance of the difference in the distribution of allele sharing between the 196R/R⫹ group and the 196R/R⫺ group.
ACKNOWLEDGMENTS
The authors are grateful to the patients and their fami-lies; to Dr. J. F. Prudhomme, Dr. C. Bouchier, and Dr. J. Weissenbach (Genethon) and Dr. M. F. Legrand, and Dr. G. Thomas (Fondation Jean Dausset-CEPH) for technical support; and to Dr. P. Brooks for critical reading of the manuscript.
REFERENCES
1. Deighton CM, Walker DJ, Griffiths ID, Roberts DF. The contri-bution of HLA to rheumatoid arthritis. Clin Genet 1989;36: 178–82.
2. Risch N. Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 1990;46:222–8.
3. Silman A. Rheumatoid arthritis. In: Silman A, Hochberg M, editors. Epidemiology of the rheumatic diseases. Oxford: Oxford University Press; 1993. p. 7–68.
4. Lawrence JS. Heberden Oration, 1969: rheumatoid arthritis— nature or nurture? Ann Rheum Dis 1970;29:357–79.
5. Aho K, Koskenvuo M, Tuominen J, Kaprio J. Occurrence of rheumatoid arthritis in a nationwide series of twins. J Rheumatol 1986;13:899–902.
6. Silman AJ, MacGregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, et al. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol 1993;32:903–7. 7. Statsny P. Mixed lymphocyte culture typing cells from patients
with rheumatoid arthritis. Tissue Antigens 1974;4:571–9. 8. Gregersen PK, Silver J, Winchester RJ. The shared epitope
hypothesis: an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987;30: 1205–13.
9. Rigby AS, MacGregor AJ, Thomson G. HLA haplotype sharing in rheumatoid arthritis sibships: risk estimates subdivided by proband genotype. Genet Epidemiol 1998;15:403–18.
10. Cornelis F, Faure S, Martinez M, Prud’homme JF, Fritz P, Dib C, et al. New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study. Proc Natl Acad Sci U S A 1998;95: 10746–50.
11. Shiozawa S, Hayashi S, Tsukamoto Y, Goko H, Kawasaki H, Wada T, et al. Identification of the gene loci that predispose to rheumatoid arthritis. Int Immunol 1998;10:1891–5.
12. Kemper O, Derre J, Cherif D, Engelmann H, Wallach D, Berger R. The gene for the type II (p75) tumor necrosis factor receptor (TNF-RII) is localized on band 1p36.2-p36.3. Hum Genet 1991; 87:623–4.
13. Cornelis F, for ECRAF. New susceptibility locus for rheumatoid arthritis on chromosome 1 [abstract]. Am J Hum Genet 1998;63: A286.
14. Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Monteiro J, et al. A genome wide screen in multiplex rheumatoid arthritis families suggests genetic overlap with other autoimmune diseases. Am J Hum Genet 2001;68:927–36.
15. Brennan FM, Maini RN, Feldmann M. TNF alpha: a pivotal role in rheumatoid arthritis? Br J Rheumatol 1992;31:293–8. 16. Husby G, Williams RC Jr. Synovial localization of tumor necrosis
factor in patients with rheumatoid arthritis. J Autoimmun 1988;1: 363–71.
17. Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleisch-mann RM, Bulpitt KJ, et al. Etanercept therapy in rheumatoid arthritis: a randomized, controlled trial. Ann Intern Med 1999;130: 478–86.
18. Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. Randomised double-blind comparison of chimeric mono-clonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet 1994;344:1105–10. 19. Bazzoni F, Beutler B. The tumor necrosis factor ligand and
receptor families. N Engl J Med 1996;334:1717–25.
20. Deleuran BW. Cytokines in rheumatoid arthritis: localization in arthritic joint tissue and regulation in vitro. Scand J Rheumatol 1996;25 Suppl 104:1–34.
21. Cope AP, Aderka D, Doherty M, Engelmann H, Gibbons D, Jones AC, et al. Increased levels of soluble tumor necrosis factor receptors in the sera and synovial fluid of patients with rheumatic diseases. Arthritis Rheum 1992;35:1160–9.
22. Steiner G, Studnicka-Benke A, Witzmann G, Hofler E, Smolen J. Soluble receptors for tumor necrosis factor and interleukin-2 in serum and synovial fluid of patients with rheumatoid arthritis, reactive arthritis and osteoarthritis. J Rheumatol 1995;22:406–12. 23. Roux-Lombard P, Punzi L, Hasler F, Bas S, Todesco S, Gallati H, et al. Soluble tumor necrosis factor receptors in human inflamma-tory synovial fluids. Arthritis Rheum 1993;36:485–9.
24. Santee SM, Owen-Schaub LB. Human tumor necrosis factor receptor p75/80 (CD120b) gene structure and promoter charac-terization. J Biol Chem 1996;271:21151–9.
25. Komata T, Tsuchiya N, Matsushita M, Hagiwara K, Tokunaga K. Association of tumor necrosis factor receptor 2 (TNFR2) poly-morphism with susceptibility to systemic lupus erythematosus. Tissue Antigens. 1999;53:527–33.
26. Beltinger CP, White PS, Maris JM, Sulman EP, Jensen SJ, LePaslier D, et al. Physical mapping and genomic structure of the human TNFR2 gene. Genomics 1996;35:94–100.
27. Shibue T, Tsuchiya N, Komata T, Matsushita M, Shiota M, Ohashi J, et al. Tumor necrosis factor ␣ 5⬘-flanking region, tumor necrosis factor receptor II, and HLA–DRB1 polymorphisms in Japanese patients with rheumatoid arthritis. Arthritis Rheum 2000;43:753–7. 28. Barton A, John S, Ollier WER, Silman A, Worthington J.
Asso-ciation between rheumatoid arthritis and polymorphism of tumor necrosis factor receptor II, but not tumor necrosis factor receptor I, in Caucasians. Arthritis Rheum 2001;44:61–5.
29. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24.
30. Dib C, Faure S, Fizame C, Samsom D, Drouot N, Vignal A, et al. A comprehensive genetic map of the human genome base on 5,264 microsatellites. Nature 1996;380:152–4.
31. Al-Ansari AS, Ollier WE, Villarreal J, Ordi J, Teh LS, Hajeer AH. Tumor necrosis factor receptor II (TNFRII) exon 6 polymorphism in systemic lupus erythematosus. Tissue Antigens 2000;55:97–9. 32. Spielman RS, McGinnis RE, Ewens WJ. Transmission test for
linkage disequilibrium: the insulin gene region and insulin-depen-dent diabetes mellitus (IDDM). Am J Hum Genet 1993;52: 506–16.
33. Rubinstein P, Walker M, Carpenter C, Carrier C, Krassner J, Falk C, et al. Genetics of HLA disease associations: the use of the haplotype relative risk (HRR) and the “haplo-delta” (Dh) esti-mates in juvenile diabetes from three racial groups. Hum Immunol 1981;3:384.
34. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001;411:599–603. 35. MacKay KR, Eyre S, Barrett J, Laval S, Myerscough A, Milicie A,
et al. Linkage analysis of potential rheumatoid arthritis non-HLA susceptibility loci [abstract]. Arthritis Rheum 2000;43 Suppl 9:S69.