Cartographie de QTLs et analyse transcriptomique : identification de gènes candidats impliqués dans la synthèse des composés phénoliques chez l’épinette
blanche
Mémoire
Justine Laoue
Maîtrise en sciences forestières - avec mémoire Maître ès sciences (M. Sc.)
Québec, Canada
Cartographie de QTLs et analyse
transcriptomique : identification de gènes
candidats impliqués dans la synthèse des composés phénoliques chez l’épinette blanche
Mémoire
Justine Laoué
Sous la direction de :
Jean Bousquet, directeur de recherche
Nathalie Isabel, codirectrice de recherche
Résumé
Les changements climatiques actuels accroissent les stress auxquels sont soumis les arbres forestiers, en modifiant l’environnement dans lequel ils évoluent au cours de leur vie. Il est donc nécessaire de mieux comprendre et mitiger leurs impacts afin de préserver la santé des forêts et maintenir leur productivité. Les composés phénoliques représentent une classe majeure de métabolites secondaires impliqués dans les mécanismes de défense chez les plantes. De précédentes études ont montré que ces composés jouaient un rôle important dans les réponses aux stress biotiques et abiotiques chez les conifères (Hammerbacher et al., 2014;
Warren et al., 2015). Dans cette étude, nous avons tiré profit des nombreuses ressources génomiques développées chez l’épinette blanche afin d’étudier les bases génomiques de la production constitutive des composés phénoliques chez cette espèce. En premier lieu, nous avons réalisé une analyse de cartographie de QTLs (Quantitative Trait Locus) portant sur la production de neuf composés phénoliques chez une descendance biparentale d’épinette blanche, et ce pour deux années distinctes. Cette analyse a permis d’identifier 17 QTLs significatifs, dont un à effet majeur chez les néolignanes expliquant jusqu’à 91,3 % de la variance phénotypique. Nous avons ensuite utilisé une approche de RNA-seq afin d’identifier des gènes différentiellement exprimés (DEGs) entre des individus présentant des phénotypes contrastés (concentrations élevées ou basses en composés phénoliques), pour les cinq métabolites chez lesquels des QTLs significatifs avaient été détectés pour les deux années de mesure. Tous métabolites confondus, 603 DEGs ont pu être identifiés, dont 50 étaient associés à la voie de biosynthèse des composés phénoliques. Dans sa globalité, cette étude a contribué à générer de nouvelles connaissances relatives aux bases génétiques des réponses aux stress biotique et abiotique chez l’épinette blanche. Ces nouvelles connaissances pourront être utilisées afin de sélectionner des arbres présentant une résilience accrue aux stress environnementaux dans le cadre de programmes d'amélioration génétique des conifères.
Abstract
Climate change is increasing the biotic and abiotic stresses faced by forest trees, altering the environment in which they evolve during their lifetime. It is therefore necessary to better understand and mitigate the impact of these stresses in order to preserve the health of forests and maintain their productivity. Phenolic compounds represent a major class of secondary metabolites involved in defense mechanisms in plants. Previous studies have shown that some of these compounds play an important role in the responses to biotic and abiotic stresses in conifers (Hammerbacher et al., 2014; Warren et al., 2015). In this study, we took advantage of the many genomic resources developed in white spruce in order to study the genomic bases of the constitutive production of phenolic compounds in this species. First, we performed QTL (Quantitative Trait Locus) mapping for nine phenolic compounds in two separate years, using a biparental white spruce progeny. This analysis identified 17 significant QTLs, including a major effect QTL explaining 91.3% of the phenotypic variance in neolignans.
We then used an RNA-seq approach to identify differentially expressed genes (DEGs) among individuals with contrasting phenolic content (high or low concentrations of phenolic compounds), for five metabolites for which significant QTLs had been detected in both years of measurement. Overall, 603 DEGs were identified, of which 50 were directly related to the biosynthetic pathway of phenolic compounds. In conclusion, this study generated new knowledge regarding the genetic basis of biotic and abiotic stress responses in white spruce.
This new knowledge can now be used to select trees with increased resilience to environmental stress within the framework of genetic improvement programs for conifers.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... viii
Liste des figures ... x
Liste des abréviations, sigles, acronymes ... xiii
Remerciements ... xv
Avant-propos ... xvi
Introduction ... 1
Effets des changements climatiques sur les forêts boréales ... 1
Stress biotiques : insectes et pathogènes ... 2
Stress abiotiques : sécheresse ... 2
Resistance aux stress environnementaux chez les conifères : les composés phénoliques... 3
Les composés phénoliques chez les conifères ... 4
Rôles des composés phénoliques dans les stress biotiques ... 5
Rôles des composés phénoliques dans les stress abiotiques ... 6
Cas particulier des néolignanes ... 7
Identification des bases génétiques responsables des variations constitutives en composés phénoliques ... 8
La cartographie de QTLs ... 8
L’analyse transcriptomique ... 11
Espèce cible : l’épinette blanche (Picea glauca) ... 12
Présentation générale ... 12
Diversité génétique de l’épinette blanche ... 14
Ressources génomiques et transcriptomiques ... 15
Programmes d’amélioration génétique de l’épinette blanche ... 16
Hypothèses et objectifs de recherche... 18
Références ... 21
Chapitre 1: Integrating QTL mapping and transcriptomics to decipher the genetic architecture of the metabolism of phenolic compounds in the conifer white spruce ... 34
1.1 Résumé ... 34
1.2 Abstract ... 35
1.3 Introduction ... 36
1.4 Material and methods ... 38
1.4.1 Workflow ... 38
1.4.2 Plant material and phenotypic data measurements ... 40
1.4.3 QTL analyses... 41
1.4.4 RNAseq experiment ... 42
1.5 Results ... 44
1.5.1 Phenotypic trait variation and correlation ... 44
1.5.2 QTL mapping analyses... 47
1.5.3 Transcriptomic analysis... 51
1.6 Discussion ... 62
1.6.1 Phenotypic data and metabolite correlations ... 62
1.6.2 Genomic architecture of phenolic compounds metabolism ... 63
1.6.3 Gene families and key genes involved in spruce phenolic compounds biosynthesis pathway ... 64
1.6.4 Functional annotations and GO analyses reveal candidate genes involved in environmental stress responses ... 67
1.6.5 Integrating QTL mapping and transcriptomic approaches: potential and limits 69 1.7 Conclusions ... 70
1.8 Acknowledgements ... 80
1.9 References ... 80
1.10 Supplementary material ... 93
1.10.1 Supplementary methods ... 93
1.10.2 Supplementary figures... 95
1.10.3 Supplementary tables ... 100
Conclusions ... 106
Principaux résultats et analyse critique ... 106
Détection des QTLs ... 106
Analyse d’expression différentielle ... 108
Applications potentielles et perspectives de recherche ... 110 Application dans un contexte d’amélioration génétique de l’épinette blanche ... 110 Carte génétique et annotation des gènes positionnés ... 112 Des approches innovantes pour identifier les bases génétiques de la biosynthèse des composés phénoliques ... 113 Vers une meilleure caractérisation fonctionnelle des gènes impliqués dans la biosynthèse des composés phénoliques ... 114 Références ... 116
Liste des tableaux
Table 1.1 Phenolic compounds analysed in the white spruce F1 full-sib family. ... 40 Table 1.2 Descriptive statistics of measured metabolites in the white spruce F1 full-sib family ... 45 Table 1.3 Summary statistics of significant QTLs (LOD score > 3.1) detected for eight phenolic compounds in the C94-1-2516 white spruce biparental family in 2014 and 2017.49 Table 1.4 List of differentially expressed genes (DEGs; adjusted p-value ≤ 0.05) involved in phenolic compounds metabolism based on their functional annotations. Sequence description: annotations obtained using BLAST2GO (p-value < 0.05); InterPro classification: most informative InterPro names. DEGs with a log2-fold change ≥ 1 were considered upregulated (labelled as UP), and DEGs with a log2-fold change ≤ -1 were considered downregulated (labelled as DOWN). Piceid does not appear in the table as none of the 3 DEGs identified for this metabolite was associated with the phenilpropanoid pathway. ... 57 Table 1.5 Differentially expressed genes also identified in QTL mapping. Piceid and procyanidin B1 do not appear in this table, as no DEGs was found within QTL regions for these metabolites. Upregulated and downregulated DEGs are referred to as UP and DOWN in the Regulation column, respectively. ... 61 Table S1.1 List and putative functions of genes identified in QTLs. Sequence description:
annotation obtained using BLAST2GO (p-value < 0.05); InterPro classification: most informative InterPro name; “NA” means that no sequence similarity was found…………100 Table S1.2 Major gene family identified in QTLs. ... 100
Table S1.3 Summary of RNA-Seq and mapping statistics. ... 101
Table S1.4 List of the 603 differentially expressed genes identified for each metabolite.
Putative functions of the DEGs are reported as follow: Sequence description: annotation obtained using BLAST2GO (p-value < 0.05); InterPro classification: most informative InterPro name; “NA” means that no sequence similarity was found. List and position of DEGs mapped on the white spruce genome are reported in this table. Among the 603 genes found, 213 could be positioned on the reference genome map (Pavy et al., 2017). ... 103 Table S1.5 List of differentially expressed genes classified into BIN functions in MapMan.
Some of the genes may be present multiple times in different bins. ... 103
Table S1.6 Picea abies and Arabidopsis thaliana homologs for the subset of candidate genes included in the improved spruce phenylpropanoid pathway presented in Figure 1.8. ... 104 Table S1.7 Candidate genes found for adaptation to climate in Hornoy et al. (2015)... 105
Liste des figures
Figure 1 Mécanismes de réponses induites chez les plantes en réponse aux stress environnementaux. Tiré de War et al. (2012). ... 4 Figure 2 Carte de distribution de l’épinette blanche (Picea glauca) au Canada.
(https://aimfc.rncan.gc.ca/fr/arbres/fiche/38) ... 14 Figure 1.1 Schematic representation of the experimental design. Bark samples were collected on 164 and 202 white spruce siblings in 2014 and 2017, respectively; and QTL analyses were performed on nine metabolites for both years. Based on the results of the QTL mapping analyses, five metabolites were retained for subsequent transcriptomic analysis.
RNA sequencing was performed on a subset of individuals displaying high versus low metabolite contents in 2017. For each five metabolites, differentially expressed genes (DEGs) among pools of individuals displaying contrasted phenotypes were identified……39 Figure 1.2 (A) Bimodal distribution of neolignans concentrations measured in August 2014 and 2017 in a white spruce biparental progeny (cross C94-1-2516; parents ♀77111 × ♂2388).
(a) Concentrations measured in 2014 for 164 descendants from the QTL mapping population C94-1-2516, (b) concentrations measured in 2017 for 202 descendants from the QTL cross C94-1-2516. The percentage reported in dotted line boxes indicate the proportion of individuals with high and low neolignans concentrations (B) Neolignans concentrations measured in August (a) 2015, (b) 2016 and (c) 2017 for the two parents of the cross ♀77111 and ♂2388 (number of clones n=3-4). ... 46 Figure 1.3 Pairwise phenotypic correlations between phenolic compounds contents measured in 2014 (bottom left) and 2017 (top right). Metabolites are classified into three major classes. The scale bar reports positive (red) and negative (blue) correlations. For each phenotypic correlation, the correlation coefficient and significance level are indicated. Levels of significance are reported as follow: * for P < 0.05, ** for P < 0.01 and *** for P < 0.001.
NS indicates non-significant correlations. ... 47 Figure 1.4 Results of differential expression analyses for five phenolic compounds. (A) Proportion of genes differentially expressed (DEGs) among pools of individuals displaying contrasted phenotypes, for each metabolite. The proportion of DEGs was calculated as the number of DEGs divided by the total number of genes expressed (Tgenes). The number of DEGs and Tgenes (in parenthesis) are reported above the bars. (B) Proportion of downregulated (log2fc < -1) and upregulated (log2fc > 1) DEGs in high meatbolite content individuals, for each metabolite. The number of regulated genes is reported above the bars.
Piceid is not presented in this panel, as no up- or downregulated DEGs were identified for this metabolite. (C) Venn diagrams showing the overlap among the 603 DEGs identified for all five metabolites. (D) Venn diagrams showing the overlap among regulated DEGs
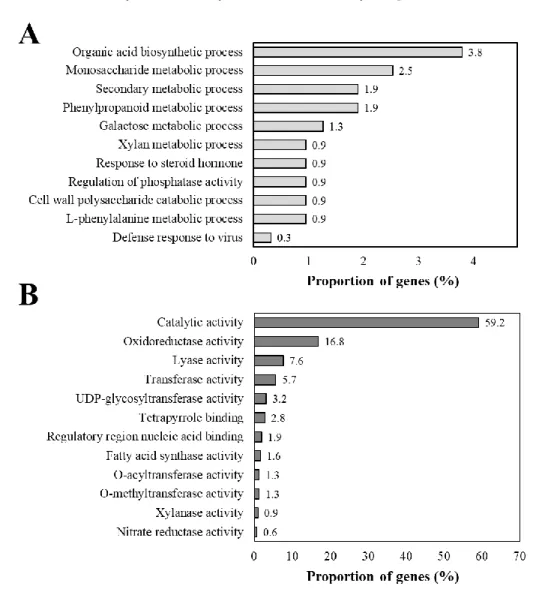
identified for the five metabolites. Piceid and Procyanidin B1 do not appear in this diagram because no overlap was found between their regulated DEGs. Since all regulated DEGs shared by gallocatechin and taxifolin were oppositely regulated (upregulated for taxifolin glucoside and downregulated for gallocatechin), the Venn diagram illustrates the overlap between upregulated taxifolin glucoside DEGs (“Taxifolin glucoside UP”) and downregulated gallocatechin DEGs (“Gallocatechin DOWN”). ... 52 Figure 1.5 GO enrichment analyses on differentially expressed genes (DEGs) identified for all five phenolic compounds. Enrichment analyses were performed with Blast2GO using Fisher’s exact tests (p-value < 0.05) for (A) biological processes and (B) molecular functions.
Relevant functional categories are presented. ... 53 Figure 1.6 MapMan-based classification of differentially expressed genes (DEGs) involved in stress responses. The scale bar represents positive (red) and negative (blue) regulation of gene expression based on log2fc scores. Black squares represent genes that were either upregulated or downregulated depending on the phenolic compound considered. (A) Expression profiles of DEGs involved in biotic stress response. Several aspects of plant biotic stress response are represented, including pathogen recognition, activation of transcription factors, or the expression of defense genes leading to the production of defense molecules.
(B) Expression profiles of DEGs involved in secondary metabolism. ... 56
Figure 1.7 Differentially expressed transcription factors (TFs) identified for all five phenolic compounds showing significant QTLs for the two years of measurement. Histograms represent the number and proportion of TFs identified as differentially expressed, classified into major families. Black bars represent the proportion of differentially expressed TFs neither up- or downregulated, and red and blue bars represent the proportion of TFs upregulated and downregulated, respectively. TFs families are presented in order of increasing abundance. For each family, the number of TFs differentially expressed, and the proportion of DEGs relative to the total number of TFs identified for the family is reported.
For LOB family, there are two differentially expressed TFs including one upregulated for the taxifolin glucoside and one downregulated for neolignans and gallocatechin. ... 59 Figure 1.8 Improved phenolic compounds biosynthesis pathway in spruce. The pathway presented here is adapted from Warren et al. (2015). Candidate gene IDs were retrieved from the gene catalog GCAT3.3 (Rigault et al., 2011) and are reported in black and italic. Genes identified in QTLs only are shown in orange boxes. The down- or up-regulation of key genes are indicated by blue and red boxes, respectively. Remaining DEGs (i.e. excluding up- and downregulated genes) are shown in white boxes. Genes within the taxifolin glucoside QTL and differentially expressed for gallocatechin are shown in white boxes with bold margins.
The metabolite for which genes are differentially expressed is reported in each box according to the following nomenclature: G, gallocatechin; N, neolignans; P, piceid; PB, procyanidin B1; Tg, taxifolin glucoside. ... 79
Figure S1.1 Distributions of metabolite concentrations for nine phenolic compounds measured in 2014 (A) and 2017 (B) in a white spruce biparental progeny (202 descendants;
cross ♀77111 × ♂2388)……….95 Figure S1.2 Heatmap of differentially expressed genes (DEGs) identified for five phenolic compounds. The levels of gene expression are presented for progeny having high (“high”) and low (“low”) phenolic content. The scale bar represents positive (red) and negative (blue) regulation of gene expression based on log2fc scores. For gallocatechin, piceid, procyanidin B1 and taxifolin glucoside 10 descendants were taken for each phenotypic category (high and low). For neolignans, 35 descendants were taken. ... 96 Figure S1.3 MapMan classification of genes differentially expressed (DEGs) among pools of descendants displaying contrasted phenotypes for five phenolic compounds. DEGs were classified into different functional categories or ‘BINs’ (according to MapMan nomenclature). The percentage of DEGs assigned to each functional category is shown in the right frame. ... 97 Figure S1.4 Up- and downregulated transcription factors (TFs) identified for gallocatechin, neolignans and taxifolin glucoside phenolic compounds according to their log2 fold-change.
Transcription factors involved in the phenolic biosynthesis pathway according to the literature are marked with a star (*)... 98 Figure S1.5 Hierarchical Gene Ontology (GO) trees for DEGs co-localizing with QTLs, based on their biological process (A) and molecular function (B) classification. ... 99
Liste des abréviations, sigles, acronymes
ANR: Anthocyanidin Reductase ANS : Anthocyanidin Synthase 4CL: 4-Coumaroyl CoA Ligase C4H: Cinnamate 4-Hydroxylase C3H: p-Coumarate 3-Hydroxylase
CAD : Cinnamyl Alcohol Deshydrogenase CCoAOMT: Caffeoyl CoA O-Methyltransferase CCR: Cynamoyl CoA Reductase
cDNA: Complementary DNA CHS: Chalcone Synthase
CYP75A: Flavonoid 3’,5’-Hydrolase DEG: Differentially Expressed Gene
DFR: Bifunctional Dihydroflavonol 4-Reductase/Flavanone 4-Reductase DIR: Dirigent Protein
DNA: Deoxyribonucleic Acid ESTs: Expressed Sequence Tags F3H: Flavanone 3-Hydroxylase FLS: Flavonol Synthase
HCT: Hydroxycinnamoyl CoA ligase/Quinate hydroxycinnamoyl transferase HPLC-MS: High Performance Liquid Chromatography-Mass Spectrometry LAR : Leucoanthocyanidin Reductase
MFFP: Ministère des Forêts, de la Faune et des Parcs of Quebec mQTL: metabolite Quantitative Trait Loci
PAL; Phenylalanine Ammonia-Lyase PLR: Pinoresinol Lariciresinol Reductase PCR: Polymerase Chain Reaction
RNA: Ribonucleic Acid
SNPs: Single Nucleotide Polymorphisms UGT: UDP-dependent glucosyl transferase
À mes grand-parents
Remerciements
J’aimerais d’abord remercier Jean Bousquet qui m’a donné l’opportunité de réaliser ce projet et m’a permis d’approfondir mes connaissances en génomique forestière. Je le remercie également pour son soutien et la confiance qu’il m’a donnée. J’aimerais également remercier Nathalie Isabel du Centre de foresterie des Laurentides pour m’avoir codirigé, ainsi que Almuth Hammerbacher du Max Planck Institute pour avoir contribué à la quantification des métabolites. Je tiens également à remercier Ilga Porth et Jean Beaulieu pour avoir accepté d’évaluer ce mémoire et avoir apporté des suggestions constructives d’amélioration du manuscrit. Un merci également à Marie-Claude Gros-Louis et toute l’équipe de terrain du CFL pour m’avoir aidé à réaliser l’échantillonnage des arbres et merci à Manuel Lamothe pour m’avoir guidé dans l’analyse des QTLs. J’aimerais également remercier toute l’équipe de professionnels de recherche et d’étudiants-chercheurs du laboratoire de la Chaire de recherche du Canada en génomique forestière. Aida Azaiez et Isabelle Giguère qui m’ont fourni une aide très précieuse pour réaliser tous les travaux en laboratoire dont ceux reliés à la transcriptomique. Sébastien Gérardi et Julien Prunier, pour leur aide en bioinformatique.
Merci infiniment pour toute votre aide dans les analyses, le labo et le reste. Sébastien Gérardi, un gros merci aussi pour tes multiples conseils lors de la rédaction du mémoire et de l’article.
Un énorme merci également à Claire Depardieu pour ton « coaching » et ton soutien moral ainsi que pour ton aide lors de la rédaction de l’article.
Et puis bien sûr, je remercie aussi tous mes camarades de bureau et du deuxième étage de l’Institut de Biologie Intégrative et des Systèmes du pavillon Marchand. Merci pour votre soutien au travail et merci pour tous les moments « hors travail » !
Merci à mes parents et à mes grands-parents qui me soutiennent dans tous mes projets.
Et pour finir, merci Alexandre pour ton soutien. Merci de me donner confiance en moi.
Avant-propos
La première partie de ce mémoire est une introduction générale comprenant une revue de littérature qui traite des principaux concepts nécessaires à la compréhension des thèmes abordés dans le reste du mémoire. Cette partie se conclut par la présentation des objectifs et hypothèses qui ont guidé le travail effectué.
Le premier chapitre est un article scientifique rédigé en anglais par l’étudiante (première auteure) et corrigé par les co-auteurs, et qui sera soumis à une revue scientifique internationale. Jean Bousquet (directeur) et Nathalie Isabel (codirectrice) ont participé à la conception du projet, à son financement et à la coordination de sa réalisation. Marie-Claude Gros-Louis a participé à la conception du design d’échantillonnage. Aida Azaiez a aidé à réaliser les travaux expérimentaux en laboratoire. Brian Boyle a participé au design de l’expérience de séquençage. Sébastien Gérardi et Manuel Lamothe ont fourni de l’aide dans les analyses bioinformatiques ainsi que de l’aide pour la rédaction. Claire Depardieu a apporté de l’aide dans la rédaction et l’interprétation des résultats au niveau physiologique.
Almuth Hammerbacher a permis l’obtention des données métaboliques qui étaient à la base même de toutes les autres analyses génomiques et transcriptomiques. L’article est lui-même composé de cinq sous-parties. La première partie est une introduction synthétique reprenant les concepts essentiels de l’introduction générale du mémoire. La deuxième partie, le matériel et les méthodes, contient l’information nécessaire pour reproduire l’étude ainsi qu’une explication détaillée des analyses effectuées. La partie suivante est composée des résultats obtenus qui sont interprétés et analysés dans l’avant-dernière partie, où l’on discute des implications de ces derniers. L’article se termine par une conclusion.
La dernière partie est une conclusion générale du mémoire. Cette partie contient les conclusions quant à l’atteinte des objectifs de l’étude et la vérification des hypothèses posées lors de l’introduction du mémoire. Elle se termine par un récapitulatif des apports de cette étude ainsi que des perspectives de recherche futures permettant d’en étendre la portée.
Introduction
Effets des changements climatiques sur les forêts boréales
Les forêts boréales abritent une importante diversité biologique qui leur confère une grande valeur écologique. Au Canada en particulier, elles possèdent également une grande valeur socio-économique et environnementale du fait des retombées économiques générées par leur utilisation (Natural Resources Canada Annual report, 2018) ou de leur capacité à emmagasiner le dioxyde de carbone (CO2) en provenance de l’atmosphère, de purifier l’air et l’eau ainsi que de régulariser le climat à grande échelle (Hogg & Bernier, 2005). La forêt boréale canadienne a une importance mondiale puisqu’elle couvre environ 28 % de la forêt boréale planétaire. Ainsi, l’aménagement de cette forêt dans le but d’en préserver ses rôles écologiques et environnementaux constitue un enjeu majeur au niveau national et même planétaire. Les forêts boréales canadiennes sont dominées par les conifères et regroupent de nombreuses espèces telles que les épinettes (Picea spp.), les sapins (Abies spp.), les pins (Pinus spp.) ou les mélèzes (Larix spp.) (Lenihan, 1993).
Cependant, comme toutes les autres forêts du monde, la forêt boréale canadienne est exposée aux changements climatiques qui apportent constamment de nouvelles menaces telles que l’introduction de pathogènes et d’insectes (stress biotiques) ou l’augmentation de la fréquence et de la magnitude des événements climatiques extrêmes tels que les sécheresses / épisodes de pluies, les tempêtes ou l’augmentation des températures (stress abiotiques) (Rustad et al., 2009). Les arbres forestiers sont par nature très sensibles aux modifications de leur environnement car ils sont sessiles et longévives. Ainsi, il est anticipé que ces derniers auront de la difficulté à faire face aux changements climatiques si le rythme auquel les changements s’opèrent actuellement se maintient (Davis, 2001). Au Canada, la perte de productivité des forêts engendre des coûts annuels très importants s’élevant à des dizaines, voire des centaines de millions de dollars, pour les dommages dus aux insectes seulement (Natural Resources Canada Annual report, 2018). Dans un tel contexte, il est donc important de comprendre les effets des stress biotiques et abiotiques sur les arbres forestiers.
Stress biotiques : insectes et pathogènes
Les insectes et les pathogènes sont les agents de perturbation les plus répandus dans les forêts nord-américaines. Ils peuvent causer des pertes annuelles moyennes en bois deux fois plus importantes que celles dues aux incendies et avoir un impact économique cinq fois plus important (Volney et Fleming, 2000). Les insectes et pathogènes entretiennent divers types d’interactions biologiques avec les espèces d’arbres forestiers, certaines d’entre elles pouvant entraîner des conséquences désastreuses (Logan et Powell, 2004). Ainsi, certains agents perturbateurs biotiques, tels que les scolytes, peuvent tuer leurs hôtes (Stark, 1982; Bentz et al., 2010), alors que d’autres, tels que les défoliateurs, les insectes racinaires ou les agents pathogènes racinaires, endommagent les arbres et les prédisposent à la mortalité par stress secondaire (Armstrong et Ives, 1995; Wallin et Raffa, 1999; Boulanger et Arseneault, 2004).
Les plus importants dommages dans les forêts sont causés par les épidémies d'insectes phytophages, dont la fréquence et l’intensité semblent liées aux changements climatiques (Dale et al., 2001). Bien que plusieurs facteurs climatiques interviennent conjointement, le réchauffement global actuel devrait avoir des effets particulièrement néfastes en provenance des épidémies (Bale et al., 2002), étant donné que le succès reproducteur des populations d'insectes est corrélé à la température saisonnière (Danks, 1987). Ainsi, une adaptation génétique rapide des insectes aux variations saisonnières de température a déjà été documentée (Balanyá et al., 2006; Bradshaw et Holzapfel, 2006), et plusieurs espèces ont étendu leurs aires de répartition suite à l’apparition de nouvelles niches favorables créées par la hausse des températures (Battistia et al., 2006; Nealis et Peter, 2009). La prolifération des insectes due aux hausses des températures ou à l’affaiblissement des mécanismes de défense des arbres peut également entraîner celle d’organismes pathogènes symbiotiques tels que les champignons, bactéries, nématodes et acariens et augmenter significativement le succès reproducteur des insectes (Hofstetter et al., 2006, Cardoza et al., 2008) en leur apportant des avantages nutritionnels et/ou défensifs (Six et Klepzig, 2004).
Stress abiotiques : sécheresse
La question de l’effet des changements climatiques sur la productivité forestière constitue un enjeu majeur, surtout dans les régions sujettes aux sécheresses fréquentes comme la partie intérieure de l’Ouest canadien (Hogg et Bernier, 2005). Malgré l’incertitude quant à la
prédiction de la direction des changements climatiques à l’échelle régionale, on sait que ces changements vont avoir des impacts directs sur la productivité, la santé et la régénération des forêts (Rustad et al., 2009). Selon les prédictions actuelles, le réchauffement global de la planète devrait entraîner une augmentation des températures mondiales moyennes sur la période 2081-2100 de plus de 1,5 ° C par rapport à la période 1850-1900, ainsi que des épisodes de sécheresse plus fréquents et intenses (Collins et al., 2013). De récentes études ont déjà montré que la mortalité forestière due à la hausse des températures et à la sécheresse avait rapidement augmenté à l’échelle mondiale au cours de la dernière décennie (van Mantgem et Schwilk, 2009; Allen et al., 2010). Ceci est d’autant plus préoccupant que les forêts boréales sont particulièrement sensibles à la sécheresse, notamment de par la nature de leurs sols souvent minces (Lenton et al., 2008). Ainsi, la mortalité des arbres associée aux sécheresses a déjà augmenté dans l’ouest de l’Amérique du Nord (van Mantgem et Schwilk, 2009), en combinaison avec des épidémies de scolytes et d’autres ravageurs (Breshears et al., 2005; Raffa et al., 2008). En effet, il a été démontré que les sécheresses étaient corrélées avec l’apparition d’épidémies d’insectes et associées à la mortalité chez les conifères (Logan et Powell, 2004). Bien que cela puisse varier en fonction de l’agent pathogène, ce phénomène serait dû au fait que les périodes de sécheresse favorisent l’éclosion des œufs tout en affaiblissant les arbres, ce qui augmenterait leur sensibilité aux attaques par les ravageurs (Bentz et al., 2010). De plus, des études ont constaté que la sécheresse observée dans certaines régions forestières du Canada affectait directement l’activité des feux de forêt dans ces régions, entraînant l’apparition d’incendies (Xiao et Zhuang, 2007).
Resistance aux stress environnementaux chez les conifères : les composés phénoliques
Au cours de leur évolution, les arbres ont développé divers moyens de défense physique et chimique en réponse aux stress biotiques et abiotiques auxquels ils sont exposés. Parmi eux, les métabolites secondaires jouent un rôle prépondérant dans les interactions des plantes avec leur environnement, et notamment dans les réponses aux stress environnementaux (Bennett et Wallsgrove 1994; War et al., 2012) (Figure 1). Ces composés organiques possèdent de multiples fonctions pas toujours bien élucidées et ils sont souvent répartis de manière différentielle entre les groupes taxonomiques au sein du règne végétal (Croteau et al., 2000).
Les métabolites secondaires sont classés en trois groupes selon leur origine biosynthétique :
les composés phénoliques, les terpènes et stéroïdes, ainsi que les alcaloïdes. Les composés phénoliques font partie des métabolites secondaires les plus répandus dans le règne végétal (Lattanzio et al., 2006) et sont notamment impliqués dans la synthèse de la lignine, une molécule commune chez toutes les plantes supérieures (Bourgaud et al., 2001). Le présent projet traitera uniquement d’un sous-ensemble de ces composés phénoliques, qui ont été précédemment rapportés comme étant impliqués dans les réponses aux stress environnementaux chez l’espèce à l’étude, à savoir les flavonoïdes, les lignanes et les stilbénoïdes (Richard et al., 2000; Hammerbacher et al., 2011, 2014).
Les composés phénoliques chez les conifères
Les composés phénoliques, aussi appelés phénols, sont des métabolites secondaires carbonés synthétisés par la voie du shikimate, ou voie des phénylpropanoides qui se divisent en plusieurs classes, dont les stilbènes, les lignanes, les flavonoïdes, les proanthocyanidines ou
Figure 1 Mécanismes de réponses induites chez les plantes en réponse aux stress environnementaux. Tiré de War et al. (2012).
les tanins (Saltveit, 2010). Ils jouent un rôle important dans la réponse aux stress environnementaux chez les plantes (Løkke, 1990), notamment dans les réactions de défense contre les herbivores et les pathogènes (Osswald et Benz, 1989; Schopf, 1989; Bennett et Wallsgrove, 1994). Les phénols sont très abondants chez les conifères chez lesquels ils sont également impliqués dans les réponses aux stress biotiques et abiotiques (Witzell et Martín, 2008; Delvas et al., 2011; Hammerbacher et al., 2014).
Rôles des composés phénoliques dans les stress biotiques
De nombreuses études réalisées sur l’épinette ont constaté un rôle de défense des composés phénoliques contre certains insectes (Brignolas et al., 1998; Ralph et al., 2006; Faccoli et Schlyter, 2007; Schiebe et al., 2012; Warren et al., 2015) et champignons pathogènes (Brignolas et al., 1995; Hammerbacher et al., 2011, 2013, 2014 et 2019). En tant qu’agent de défense contre les insectes, ils vont par exemple s’accumuler dans certaines parties de la plante et agir comme toxine pour les insectes qui se nourrissent de ces parties (Harborne, 2001). En effet, la première défense contre un agent pathogène ou un herbivore va être la barrière physique. Chez les conifères cela comprend l’écorce extérieure composée principalement de cellules lignifiées et subérisées. L’écorce interne et le phloème secondaire contiennent quant à eux une variété de types de cellules liées à la défense tels que le sclérenchyme fortement lignifié, le parenchyme polyphénolique, qui serait impliqué dans la production et le stockage de composés phénoliques toxiques, ainsi que les conduits de résine qui stockent les terpénoïdes (Franceschi et al., 2005; Bohlmann, 2008). En cas de blessure ou d’attaque par les herbivores, les tissus du tronc ou des branches du conifère répondent à la formation de lésions (réponse hypersensible) par la mort cellulaire et l’accumulation de composés phénoliques (Lieutier, 2002). Une étude de Franceschi et al. (1998) a démontré l’importance des cellules du parenchyme axial, aussi appelé parenchyme phénolique, dans la synthèse, le stockage et la modification des composés phénoliques en réponse à une blessure chez l’épinette de Norvège (Picea abies). Les composés phénoliques sous la forme de lignine contribuent à la formation du sclérenchyme qui permet de donner une résistance mécanique.
La lignine, en fournissant une rigidité et une hydrophobie aux parois cellulaires du xylème, permet de réduire l'alimentation par les herbivores et diminue également le contenu
nutritionnel de la feuille. La synthèse de la lignine, induite par l’attaque d’un insecte (ou d'autres agents pathogènes), va également permettre de réduire la fécondité des herbivores (Johnson et al., 2009). Une étude chez l’épinette de Norvège a également rapporté que les cellules scléreuses contenaient, en plus de la lignine, d’autres composés phénoliques tels que les stilbènes et les flavonoïdes (Li et al., 2007), suggérant que les cellules scléreuses peuvent être impliquées dans des mécanismes de défense à la fois chimique et physique. Ainsi, dans l’écorce de l’épinette, l’augmentation de la densité des cellules scléreuses a notamment été associée à une plus grande résistance au charançon (King et al., 2009). Les composés phénoliques ont également un rôle dans les mécanismes de défense contre les stress abiotiques, tels que décrit à la section suivante.
Rôles des composés phénoliques dans les stress abiotiques
Les métabolites secondaires peuvent également être impliqués dans la tolérance aux stress abiotiques tels que la sécheresse (Akula et Ravishankar, 2011). La sécheresse provoque souvent un stress oxydatif qui entraîne une augmentation de la concentration de composés phénoliques comme les flavonoïdes et les acides phénoliques présents dans les parties ligneuses des plantes telles que l’écorce et la tige (Larson, 1988). Dans ce cas de figure, les composés phénoliques agissent alors comme antioxydants (Kähkönen et al., 1999;
Siddhuraju et Becker, 2003; Maffei et al., 2007). L’effet de la sécheresse sur les composés phénoliques tels que la lignine est très peu connu, que ce soit chez les conifères ou chez les plantes agricoles (Moura et al., 2010). Cependant, certaines études ont permis de mettre en évidence que les tissus de plus en plus lignifiés au fil des saisons de croissance devenaient moins vulnérables aux potentielles embolies créées lors d’un stress hydrique (Choat et al., 2008). Les lignines fourniraient une rigidité et une résistance à la compression aux trachéides du xylème (Niklas, 1992; Chabannes et al., 2001; Herbette et al., 2015), augmentant ainsi leur résistance à la cavitation. Une étude d’Awad et al. (2012) a également montré qu’une diminution de la teneur en lignine du xylème des peupliers transgéniques augmentait leur vulnérabilité à la cavitation, mettant ainsi en évidence le rôle des lignines dans la résistance à la cavitation chez les arbres. Une autre étude de Coleman et al. (2008) chez le peuplier a montré que l'inhibition de la lignification des parois cellulaires réduisait leur conductivité
hydraulique et augmentait la sensibilité à la rupture de la paroi et à la cavitation. Ainsi, le métabolisme de la lignine serait impliqué dans la sensibilité à la cavitation et jouerait un rôle important dans la résistance des plantes aux stress hydriques.
Cas particulier des néolignanes
De précédents travaux (données non publiées constituant la base de ce projet) sur une famille d’épinette blanche provenant du Québec ont permis de mettre en évidence des concentrations en néolignanes fortement contrastées parmi les descendants à l’étude. En effet, ces derniers possédaient le plus souvent soit de fortes ou de faibles concentrations en néolignanes. Cette distribution est généralement caractéristique des traits quantitatifs contrôlés par un ou quelques gènes, ce qui en fait un trait particulièrement intéressant à étudier.
Les lignanes et les néolignanes sont un large groupe de composés phénoliques très répandus dans le règne végétal (Teponno et al., 2016). Alors que les lignanes et les néolignanes font l'objet de recherches intensives en raison de leurs activités bénéfiques pour la santé animale en tant qu’antioxydant (Kim et al., 2010), leur fonction dans le métabolisme des plantes demeure peu documentée. On leur attribue le plus souvent un rôle dans la défense contre les agents pathogènes (Davin et al., 2008; Naoumkina et al., 2010). Ils ont ainsi été décrits comme agent antifongique (Hwang et al., 2010), comme ayant une activité cytotoxique (Syu et al., 2004), et comme réduisant la mue et induisant la mortalité chez les insectes (Cabral et al., 1999, 2000). Il est probable que les différences signalées entre les effets des lignanes et des néolignanes s'expliquent non seulement par leurs spécificités structurelles, mais aussi par leur état « redox ». Par exemple, les formes réduites de composés phénoliques présentent généralement des activités anti-oxydantes, alors que leurs homologues oxydés sont cytotoxiques (Lattanzio et al., 2006). Cependant, malgré l’abondance de données in vitro soutenant l'activité antioxydante des lignanes et des néolignanes, il n'existe aucune preuve directe à ce jour d'une telle fonction in vivo. Chez les conifères, une étude de Wainhouse et al. (1990) a démontré que la survie et le développement des larves de l’insecte Dendroctonus micans étaient réduits lorsqu’elles se nourrissaient de l’écorce hautement lignifiée des épinettes, suggérant un rôle de défense des lignanes et potentiellement des néolignanes. Afin
de caractériser le rôle précis de ces composés phénoliques, il apparaît donc nécessaire de développer des connaissances sur les bases génétiques de leur synthèse.
Identification des bases génétiques responsables des variations constitutives en composés phénoliques
La compréhension des mécanismes génétiques qui gouvernent la synthèse des phénols et déterminent leurs variations constitutives au sein des individus constitue un enjeu majeur chez les plantes longévives telles que les conifères en raison de la résistance aux stress environnementaux qu’ils confèrent. D’un point de vue pratique, ces connaissances pourraient être intégrées aux outils génomiques préexistants mis en œuvre dans le cadre de programmes de conservation ou d’amélioration des arbres visant à mitiger les effets des changements climatiques actuels. L’intérêt principal de ce type d’outils, telle que la sélection génomique (voir la section « Programmes d’amélioration génétique de l’épinette blanche » pour plus de détails), est de réduire significativement la durée des cycles d’amélioration, qui peuvent nécessiter plusieurs dizaines d’années dans le cas des méthodes traditionnelles basées sur la mesure des phénotypes d’intérêt (Grattapaglia et Resende, 2011).
La cartographie de QTLs
Il existe de nombreuses méthodes permettant d’identifier les gènes ou régions génomiques impliqués dans le contrôle génétique de caractères phénotypiques d’intérêt. La cartographie d’association génétique et la cartographie de locus de caractères quantitatifs, plus communément appelée cartographie de QTLs (Quantitative Trait Locus), sont probablement les deux types d’approches les plus utilisées pour y parvenir. Toutefois, leur application dépend du type de population utilisée. Dans le cas de la cartographie d’association, on recherche des associations génotype - phénotype au sein de populations naturelles (idéalement non-structurées) ou de populations domestiquées composées d’individus faiblement apparentés. Étant donné que le déséquilibre de liaison est généralement faible dans ces populations (e.g. Pavy et al., 2012 pour les épinettes), ce type d’approche possède une très bonne résolution permettant l’identification de gènes, voire de polymorphismes,
causaux. En revanche, il est souvent difficile de détecter des effets significatifs lorsque les traits phénotypiques étudiés sont contrôlés par de multiples gènes à faibles effets (e.g.
Beaulieu et al., 2011; Lamara et al., 2016). La cartographie de QTLs repose sur le même principe d’identification d'associations génotype - phénotype mais recourt à l’utilisation de populations issues de croisements dans lesquelles les individus sont fortement apparentés (Collard et al., 2005). Par conséquent, le déséquilibre de liaison est très fort, ce qui mène généralement à la découverte de grandes régions génomiques significatives (QTLs). En revanche, les résultats obtenus ne s’appliquent qu’à la famille à l’étude et peuvent varier selon les sites dans lesquels sont établis les arbres, en raison des potentielles intéractions génotype x environnement. La suite de cette partie se focalisera plus particulièrement sur les approches de cartographie de QTLs car la population à l’étude est une descendance biparentale issue d’un croisement.
La cartographie de QTLs permet de caractériser l’architecture génomique d’un caractère quantitatif en identifiant, sur une carte génétique, les régions du génome contenant des gènes responsables de la variation du caractère observée au sein d’une descendance. Pour ce faire, on teste s’il existe des associations significatives entre les variations de type génétique et phénotypique observées chez les descendants. Les régions présentant des associations génotype – phénotype significatives parmi les descendants du croisement sont dites QTLs et il est possible de calculer le pourcentage de variance phénotypique expliqué par le QTL afin de déterminer s’il a un effet majeur ou mineur sur le phénotype. La résolution de l’analyse dépend principalement de la densité de la carte génétique qui sert à cartographier les QTLs (i.e. le nombre de marqueurs / gènes positionnés) et du nombre de descendants testés (Li et al., 2010). Ainsi, plus la carte est dense et plus le nombre de descendants testés est élevé, plus la délimitation de la région génomique sera précise.
Les analyses de QTLs ont été parmi les premières approches moléculaires utilisées pour décrire les bases moléculaires de la variation des caractères quantitatifs chez les espèces forestières (e.g. Groover et al., 1994; Kaya et al., 1999). Cependant, les analyses de QTLs présentent certaines limites chez les conifères. En général, la résolution des analyses QTLs déjà publiées est faible, dû au fait que les conifères possèdent des génomes extrêmement
grands qui rendent la production de cartes génétiques denses difficile et dispendieuse. Ainsi, les QTLs identifiés sont souvent de grande taille (plusieurs cM : Pelgas et al., 2011; Lind et al., 2014) et englobent un grand nombre de gènes, certains d’entre eux pouvant être liés à des gènes causaux sans être eux-mêmes impliqués dans le contrôle génétique du caractère d’intérêt. De plus, il est possible que les QTLs identifiés contiennent des gènes causaux qui ne sont pas encore cartographiés. Ainsi l’analyse de QTLs permet de dresser une liste de gènes candidats dont il est souvent nécessaire de valider l’implication dans le contrôle génétique du caractère afin d’interpréter les résultats obtenus. Une façon d’y parvenir est de vérifier si la fonction des gènes candidats est cohérente avec un potentiel effet des gènes sur le phénotype mesuré. Cependant, l’annotation fonctionnelle des gènes candidats n’est pas toujours connue ou précise, particulièrement chez les plantes non-modèles tels que les conifères qui, en outre, possèdent des gènes spécifiques n’existant pas chez les espèces modèles angiospermes. Un autre moyen permettant de valider les gènes candidats détectés en analyse de QTLs est d’utiliser conjointement d’autres méthodes ou types de données afin de tester les gènes candidats de façon individuelle. La plupart des méthodes alternatives, tels que les tests d’association, nécessitent d’analyser une population d’individus faiblement apparentés et ne peuvent donc pas être appliquées à une descendance de croisement. En revanche, il est possible de recourir à la transcriptomique, soit l’étude du niveau d’expression des gènes, pour tester les gènes individuellement, et ce indépendamment du type de population à l’étude. C’est donc l’approche qui a été retenue dans le cadre de l’étude qui sera présentée dans la première partie de ce mémoire.
Dans le cadre de ce projet, le caractère quantitatif mesuré est la quantité de composés phénoliques produits par les arbres. Ainsi, l’analyse devrait permettre d’identifier des QTLs associés à des variations constitutives d’abondance de différents métabolites chez les descendants mesurés, et de dresser la liste des gènes localisés dans les QTLs, qui seront considérés comme candidats. À notre connaissance, aucune étude n’a tenté d’identifier des QTLs reliés à la synthèse des composés phénoliques chez les conifères. Chez l’épinette, seule une étude de Porth et al. (2011) a permis de mettre en évidence des régions génomiques impliquées dans la voie de biosynthèse des phénylpropanoïdes par une approche de eQTL (expression de gènes reliés à la voie des phénylpropanoïdes) et de pQTL (phénotype de
croissance et susceptibilité à un insecte). En revanche chez les arbres, une étude sur l’espèce modèle Populus (nigra, deltoides, trichocarpa) a permis d’identifier des QTLs basés sur le profil métabolique des individus et ainsi détecter des régions génomiques et des potentiels gènes qui contrôleraient la synthèse de flavonoides lors de certaines étapes enzymatiques clés (Morreel et al., 2006). Ces travaux ont donc montré qu’il était possible de mener des analyses de QTLs afin d’élucider les bases génétiques de la synthèse des phénols chez les arbres et que cette approche était appropriée pour identifier les enzymes clés dans une voie métabolique complexe de composés étroitement apparentés.
L’analyse transcriptomique
L’analyse transcriptomique vise à caractériser la nature et la quantité de transcrits présents chez un individu à un instant t et dans une condition donnée. Elle renseigne donc sur le niveau d’expression des gènes dans le ou les tissus échantillonnés. Il existe plusieurs méthodes permettant de quantifier les ARN messagers au sein d’un échantillon, comme la PCR en temps réel (qPCR), les biopuces à ADN complémentaire (microarrays) ou le séquençage d’ARN (RNA-Seq). Cependant, le RNA-Seq est sans contredit la méthode la plus puissante car elle permet simultanément de séquencer l’ensemble des gènes exprimés dans un échantillon et de mesurer leur niveau d’expression. Le RNA-Seq fait partie des techniques de séquençage de nouvelle génération (NGS) qui se sont répandues récemment suite à la baisse des coûts de séquençage. Le principe général consiste à extraire les ARNs présents dans un échantillon et de les convertir en ADN complémentaire, qui seront ensuite séquencés via une technologie de séquençage à haut-débit (e.g Illumina) générant un grand nombre de petites lectures (reads) qui sont par la suite assemblées en groupes représentatifs des gènes exprimés (Wang et al., 2009). Cette technique peut ainsi être utilisée afin d’identifier des gènes différentiellement exprimés entre des tissus, des individus ou des groupes d’individus.
Bien développée chez les plantes modèles telles qu’Arabidopsis thaliana (Gan et al., 2011;
Klepikova et al., 2016), cette approche a récemment été utilisée chez les arbres afin d’identifier des transcrits exprimés de manière différentielle dans des conditions particulières. Par exemple, chez le sapin blanc (Abies alba), les analyses d'expression différentielle ont permis d’identifier des gènes candidats impliqués dans la résistance à la
sécheresse (Behringer et al., 2015). Chez le pin blanc (Pinus monticola), les analyses de RNA-Seq ont permis d’identifier des gènes potentiellement impliqués dans la résistance à l’infection par le champignon pathogène Cronartium ribicola, causant la rouille vésiculeuse du pin blanc (Liu et al., 2013).
La présente étude se concentrera sur l’identification de gènes différentiellement exprimés chez des individus présentant des variations constitutives d’abondance de métabolites, soit des individus produisant de grandes quantités de métabolites vs des individus en produisant peu. Cette analyse devrait donc permettre d’identifier des gènes impliqués dans la synthèse des composés phénoliques chez les conifères car, à notre connaissance, aucune étude de ce type n’a été réalisée chez les conifères à ce jour. En revanche, une approche similaire a par exemple été employée avec succès chez la canne à sucre (Saccharum spp.). Les auteurs ont utilisé la technique de RNA-Seq pour étudier les transcrits différentiellement exprimés entre des tissus de la canne à sucre dont la teneur en fibres variait. Les résultats ont révélé des gènes cibles pour des recherches ultérieures sur la régulation du métabolisme de la cellulose et de la lignine (Kasirajan et al., 2018). De même, de nombreuses études sur l’expression des gènes ont été conduites chez le genre Picea concernant la croissance (Stasolla, 2004;
Friedmann et al., 2007), les propriétés du bois (Pavy et al., 2008), les stress biotiques (Ralph et al., 2006; Lippert et al., 2007) et les stress abiotiques (Fossdal et al., 2007). Comprendre les mécanismes génétiques gouvernant la synthèse des phénols chez les plantes est un défi difficile compte tenu de la grande diversité de ces composés. L’usage d’outils génomiques en combinaison avec le profilage en métabolites devrait permettre d’identifier efficacement des gènes clés impliqués dans leur synthèse.
Espèce cible : l’épinette blanche (Picea glauca)
Présentation générale
L’épinette blanche (Picea glauca [Moench] Voss, famille des Pinaceae) est une espèce coniférienne originaire des forêts tempérées et boréales d’Amérique du Nord. L’épinette blanche possède une aire de répartition transcontinentale couvrant un vaste territoire compris entre l’Alaska à l’ouest et Terre-Neuve à l’est du Canada, et s’étendant des états frontaliers
des États-Unis et du sud du Canada jusqu’à la limite des arbres au nord (Figure 2). Elle pousse normalement à une hauteur de 15 à 30 m et peut atteindre 40 m de hauteur avec un diamètre du tronc de 60 cm à 200 ans. Cette espèce pousse dans des sols variés et supporte une vaste gamme de conditions climatiques (Nienstaedt et Zasada, 1990). Elle est extrêmement résistante aux basses températures allant jusqu’à -50 °C. Au Canada, elle est beaucoup utilisée pour la production de pâte à papier et de bois d’œuvre, représentant ainsi un important atout pour l’industrie forestière canadienne (Farrar et Service canadien des forêts, 1995). Avec l’épinette noire, elle constitue l'une des espèces les plus couramment plantées au Canada, représentant un total de 150 millions de semis plantés annuellement, dont plus de 25 millions sont produits dans les pépinières québécoises (Beaulieu et al., 2009).
L’épinette blanche est une espèce qui possède une grande diversité génétique (Li et al., 1997;
Namroud et al., 2008) et elle est l’un des conifères « modèles » les plus étudiés dans le domaine de la génomique. Des programmes d’amélioration génétique exploitant cette grande diversité ont été mis en place depuis les années 60 dans différentes parties du Canada et des États-Unis (Mullin et al., 2011; Lenz et al., 2013). Ces programmes étaient essentiellement centrés sur les caractères de croissance et de qualité du bois. En raison des changements climatiques actuels, un intérêt grandissant est placé dans l’étude des mécanismes génomiques de résistance aux stress environnementaux, comme en témoigne le projet Spruce-Up, un grand projet national de génomique des épinettes supporté par Génome Canada et Génome Québec, et dont l’objectif est d’apporter des connaissances, entre autres, sur la résistance aux attaques par les insectes et à la sécheresse chez cette espèce.
Diversité génétique de l’épinette blanche
L’épinette blanche possède une grande amplitude écologique comme en témoigne sa grande répartition géographique et sa capacité à croître dans des conditions climatiques variées, dont des climats et des sols extrêmes. Elle possède ainsi une grande diversité génétique lui permettant de s’adapter à des conditions environnementales variées. La variation génétique gouvernant les traits d’intérêt économique ou écologique est importante à caractériser afin d’optimiser les programmes de conservation de la diversité génétique et les programmes d’amélioration génétique. De précédentes études ont donc révélé que la structure des populations de nature historique était très faible dans l’est du Canada (Namroud et al., 2008;
de Lafontaine et al., 2010; Hornoy et al., 2015), mais qu’il existait des variations génétiques significatives entre les provenances du Québec, ainsi qu’entre les familles au sein des provenances pour des caractères de qualité du bois, pour la croissance et pour la phénologie (e.g. Li et al., 1997; Jaramillo-Correa et al., 2001). Cette tendance suggère que les provenances sont adaptées localement à leurs conditions environnementales et qu’il est donc possible d’utiliser la variation génétique préexistante au sein de l’espèce pour améliorer la performance des arbres.
Figure 2 Carte de distribution de l’épinette blanche (Picea glauca) au Canada. (https://aimfc.rncan.gc.ca/fr/arbres/fiche/38)
Ressources génomiques et transcriptomiques
L’assemblage et l’annotation de la séquence génomique et la caractérisation fonctionnelle du transcriptome des espèces constituent le socle de la compréhension générale de leur architecture génomique et de leur évolution. Le développement des ressources génomiques entrepris chez l’épinette blanche durant les deux dernières décennies en fait aujourd’hui un modèle en génomique des conifères (Warren et al., 2015). En effet, la première séquence du génome complet de l’épinette blanche a été publiée par Birol et al. (2013) et a rapidement été suivie par la publication d’une séquence améliorée deux ans plus tard (Warren et al., 2015).
Cependant, malgré les efforts considérables déployés en termes de séquençage et d’assemblage, la séquence produite demeure encore partielle et largement fragmentée. Ces lacunes sont essentiellement dues à la très grande taille du génome de l’épinette blanche (~20 Gb, Ohri et Khoshoo, 1986) et à la forte proportion d’éléments répétés, qui rendent l’assemblage de la séquence génomique complexe (Rigault et al., 2011). Afin de contourner ces difficultés, la majorité des efforts de séquençage ont d’abord été concentrés sur le transcriptome (Pavy et al., 2005). L’étude de Pavy et al. (2005) a ainsi permis de comparer les séquences codantes de l’épinette avec celles d’Arabidopsis, du riz, du peuplier et du pin afin de les annoter. Les résultats ont montré que 70% des séquences de l’épinette avaient des similarités avec Arabidopsis, le riz et le peuplier et 84% avec le pin. Ces premières analyses ont permis d’identifier des gènes candidats en vue de réaliser des analyses fonctionnelles et des études d’association. Grâce au développement du « Next Generation Sequencing » (Metzker, 2010), le catalogue de gènes de l’épinette blanche a pu être étendu à plus de 27,000 clusters, pour la plupart annotés (Rigault et al., 2011). En conjonction avec le développement des techniques de génotypage (puce à oligonucléotides), ces progrès ont également permis de générer des registres de polymorphismes nucléotidiques (SNPs, Pavy et al. 2006; 2008;
2013a) localisés dans les gènes et de relier cette variation génétique à divers facteurs environnementaux (e.g. températures, précipitation) et traits phénotypiques d’intérêt (e.g.
phénologie, productivité, qualité du bois) (e.g. Namroud et al., 2008; Beaulieu et al., 2011;
Pelgas et al., 2011; Prunier et al., 2011; Pavy et al., 2013b; Hornoy et al., 2015; Lamara et al., 2016; 2018). Ces avancées ont aussi permis de caractériser l’architecture génomique de l’épinette blanche et d’apporter des réponses quant à l’évolution de son génome en la
comparant avec celle d’autres espèces (e.g. Pavy et al., 2012; 2013b; 2017). Finalement, les données de génotypage ont permis la construction d’une carte génétique dont la résolution n’a cessé de s’améliorer au fil du temps (Pavy et al., 2008; 2012; 2017). Dans sa version la plus récente (Pavy et al., 2017), la carte génétique de l’épinette blanche inclut environ 9,000 gènes positionnés avec grande précision. L’obtention de la première carte génétique a ouvert la voie à la production d’une étude de QTLs sur le débourrement et la formation des bourgeons, ainsi que la croissance en hauteur (Pelgas et al., 2011). Bien qu’à ce jour, peu d’études de cartographie de QTLs aient été publiées en lien avec les caractères de résistance aux stress environnementaux chez l’épinette blanche (mais voir Porth et al., 2012), une étude récente deD'Odorico et al. (2020) a permis de développer une approche économique de phénotypage à haut débit via l’utilisation de drones, permettant de caractériser rapidement l'état physiologique de milliers d’arbres au champ. L’acquisition de données phénotypiques à grande échelle devrait ainsi faciliter la réalisation des études de cartographie de QTLs en lien avec les stress environnementaux chez les espèces d’arbres forestiers.
Programmes d’amélioration génétique de l’épinette blanche
Les programmes d’amélioration génétique des espèces forestières ont pour objectif d’identifier des individus et des familles démontrant des performances supérieures à la moyenne de la population dont ils sont originaires. Historiquement, les programmes d’amélioration génétique de l’épinette blanche au Canada se sont concentrés principalement sur les traits de croissance et de qualité du bois (Beaulieu et al., 2009; Mullin et al. 2011), soit des traits d’intérêt pour la production de bois. Cependant, en raison des changements climatiques actuels, les programmes d’amélioration génétique doivent désormais intégrer des nouveaux caractères tels que la résistance aux stress biotiques (e.g. insectes, pathogènes) et abiotiques (e.g. hausse des températures, sécheresses) nécessitant des actions à court terme.
Les méthodes traditionnelles d’amélioration des arbres basées sur la mesure des caractères phénotypiques sont longues (plusieurs dizaines d’années chez les espèces longévives à faible croissance comme les épinettes) et coûteuses. Fort heureusement, le développement d’outils génomiques telle que la génétique d’association ou la sélection génomique est entrain de réduire considérablement la durée des cycles d’amélioration ainsi que les coûts associés à
l’évaluation de la performance des arbres sélectionnés, tout en offrant une grande versatilité en facilitant la sélection multicritère (Lenz et al., 2020a).
Le principe des méthodes d’association consiste à identifier des marqueurs (SNPs le plus souvent) significativement associés à un caractère favorable et à sélectionner les individus sur la base de la présence ou l’absence des marqueurs plutôt que sur la mesure du caractère lui-même. Chez l’épinette blanche, l’étude de Mageroy et al. (2015) a par exemple montré que l'expression du gène de la β-glucosidase, un biomarqueur, était fortement associée à la résistance naturelle de l’épinette blanche contre la tordeuse des bourgeons de l’épinette (Choristoneura fumiferana), ouvrant la voie à la sélection rapide d’arbres plus résistants sur la base de ce biomarqueur. La sélection génomique consiste quant à elle à couvrir le génome complet de marqueurs génétiques (anonymes ou pas) et, à l’aide des profils génomiques ainsi générés et de données phénotypiques prises sur les mêmes arbres, de construire un modèle multilocus permettant de prédire un trait dans une population d’amélioration. L’approche requiert donc de construire des modèles de prédiction des phénotypes à partir des génotypes et d’estimer la précision des modèles prédictifs. Une fois le modèle développé, le processus de sélection de nouveaux arbres peut débuter aussitôt que les arbres sont génotypés, ce qui peut être fait dès leur plus jeune âge. L’utilisation de cette approche possède des avantages non négligeables pour les programmes d’amélioration des arbres forestiers. En effet, elle permet de réduire substantiellement la durée des cycles d’amélioration puisqu’il n’est plus nécessaire d’attendre de nombreuses années afin de mesurer les caractères d’intérêt (Grattapaglia et Resende, 2011; Park et al., 2016). Puisqu’elle implique des marqueurs génétiques répartis sur l’ensemble du génome, elle ne nécessite pas l’identification d’associations génétiques significatives qui, trop souvent, n’expliquent qu’une faible partie de la variance phénotypique observée. De plus, elle permet une meilleure précision pour la sélection de caractères à faible héritabilité et pour les caractères difficilement mesurables tels que la résistance aux insectes ou à la sécheresse (Heslot et al., 2012; Lenz et al., 2020a).
Motivés par la rentabilité économique de l’approche (Chamberland et al., 2020), les premiers modèles de sélection génomique ont été développés pour des caractères de croissance et de qualité de bois dans diverses populations d’amélioration d’épinette blanche du Québec (Beaulieu et al., 2014a; 2014b; Lenz et al., 2020b).
Hypothèses et objectifs de recherche
Problématique :
Les épinettes constituent la plus importante ressource forestière du Canada en produisant du bois et des fibres de haute qualité utilisés dans l'industrie. Espèces dominantes des forêts canadiennes, elles fournissent des services écosystémiques essentiels aux niveaux local et mondial. Afin de pouvoir répondre à la demande en bois d’épinette, des programmes d’amélioration génétique en Ontario, en Colombie-Britannique, en Alberta, au Nouveau- Brunswick et au Québec fournissent des stocks améliorés d'épinettes destinés à la production de semences et de plants. Cependant, les programmes d’amélioration doivent garantir productivité, qualité du bois et santé des écosystèmes, en plus de devoir faire face aux nouveaux défis posés par les changements climatiques actuels. L’utilisation d’outils génomiques rapides et efficaces devient donc de plus en plus critique afin de répondre à ces multiples contraintes. Le présent projet s’inscrit dans le projet de grande envergure Spruce- Up, qui a pour objectifs 1) de documenter l’impact socio-économique de l’intégration de la génomique dans les programmes d’amélioration génétique, 2) d’améliorer l’assemblage, le séquençage et l’annotation des génomes d’épinettes, 3) de comprendre les mécanismes génétiques et physiologiques des processus liés à la résistance aux stress biotiques et abiotiques et 4) de développer et évaluer des modèles de sélection génomique pour des applications à court terme. Les travaux présentés s’inscriront dans le cadre du troisième objectif énoncé et concerneront plus spécifiquement l’étude des mécanismes génétiques gouvernant la synthèse des composés phénoliques, des métabolites secondaires impliqués dans la résistance aux stress biotiques et abiotiques, chez l’épinette blanche. L’approche globale proposée ici pour y parvenir, qui consiste à effectuer un balayage génomique et transcriptomique complet, représente une première en ce qui a trait à la synthèse des composés phénoliques chez les conifères.
Objectif général de l’étude :
L’objectif de ce projet de maîtrise était d’étudier les bases génétiques responsables des variations constitutives en composés phénoliques observées au sein d’une descendance biparentale d’épinette blanche. Le travail effectué se divise en deux volets interreliés, soit 1)