Decipher in situ Signaling and Complex Genetics with Cellular Recording and Combinatorial
Perturbations MASSACHUSETS
OF TECHNOL
by
MAR
0
7 2
Cheryl H. Cui
LIBRARI
B. A. Sc. Engineering Science, Biomedical Engineering, University of Toronto, 2012
SUBMITTED TO THE HARVARD-MIT PROGRAM OF HEALTH SCIENCES AND
TECHNOLOGY IN PARTIAL FULFILLMENT OF THE REQUIREMENT FOR THE
DEGREE OF
DOCTORAL DEGREE IN MEDICAL ENGINEERING AND MEDICAL PHYSICS
AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
FEBURARY 2017
CMassachusetts Institute of Technology 2017. All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publicly paper and
electronic copies of this thesis document in whole or in part in any medium now known or hereafter
created.
Signature of Author:
Certified by:
Accepted by:
Signature redacted
Harvard-MI' Program in Health Sciences and Technology
Signature redacted_
Timothy K. Lu, Ph.D. Associate Professor, Department of Biological Engineering
Thesis Supervisor
Signature redacted
INSTITUTE OGY017
ES
I
Emery N. Brown, MD, Ph.D.Professor oP onputational Neuroscience and Health Sciences and Technology Director, Harvard-MIT Program in Health Sciences and Technology
Decipher in situ Signaling and Complex Genetics with Cellular Recording and Combinatorial Perturbations
by
Cheryl H. Cui
Submitted to the Harvard-MIT Division of Health Science and Technology in partial fulfillment of the requirement for
the doctoral degree in medical engineering and medical physics
Abstract
The complex, dynamic, and responsive behavior of cells arises from integrated signaling pathways and regulatory networks. With advancement in our ability to engineer mammalian cells, we harness a novel set of molecular tools to develop synthetic biology-enabled applications that help facilitate our understanding of complex biological networks and cellular behaviors. The recent discovery of the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated (Cas) system from prokaryotic adaptive immune system demonstrated unprecedented genome editing efficiency and programmability in sequence specific genome editing of mammalian cells. In this thesis, I utilized the CRISPR-Cas system to construct a combinatorial genetic perturbation platform that enables massively parallel high throughput screening of multiple gene elements. This technology platform allows systematically interrogation of higher-order interactions of genetic regulators. The later part of the work described the establishment of a genomically encoded cellular recorder with the ability to longitudinally track and record molecular events in live animals. This cellular recorder encodes cellular memory through the quantitative accumulation of targeted genomic mutations, that allows mapping of a dynamical set of gene regulatory events without the need for continuous cell imaging or destructive sampling. Together, we envision these sets of technology and tools will offer new insights into cellular process in disease and in health.
Thesis Supervisor: Dr. Timothy K. Lu
Acknowledgement
So many people helped me along the way that it would be impossible to mention them all. But I must say special thanks to my Ph.D. supervisor, Prof. Timothy Lu, my thesis committee chair, Prof. James Collins and thesis member, Prof. Alex Shalek. Many of the work described here are the results of close collaboration with colleagues and friends in the lab, Dr. Samuel Perli, Dr. Alan Wong and Dr. Gigi Choi. I also deeply appreciate the support from my graduate department, especially from Dr. Julie Greenberg, Traci Anderson and Laurie Ward. Moreover, it is impossible for me to complete this work without the foundation that my previous research supervisors has helped me established earlier on in my studies including Dr. William Stanford, Dr. Jeffrey Karp, Dr. Weian Zhao, Dr. Kevin Kain and Dr. Timothy Padera.
Graduate study is a long journey and I am fortunate to have met and became friends with some of the most brilliant minds at MIT and at Harvard including my classmates at the HST program. I especially like to thank my dearest friends, Wen Xu, Shuo Wang and Lichen Liu, whose support helped me through difficult times during my graduate studies. I also sincerely appreciate the support from my parents and my family.
Table of Contents
Abstract...
2
Acknowledgem ent ...
3
Table of Contents ...
4
List of Figures...7
1. G eneral Introduction...
10
2. Massively parallel combinatorial genetic perturbation screening ...
11
2.1 Combinatorial genetics in modern biology ...
11
2.2 Current approaches in studying combinatorial genetics ...
12
2.3 Genome engineering tools as a gene perturbation platform ...
13
3. Massively parallel combinatorial genetic perturbation screening with CRISPR-Cas9
in hum an cells...
16
3.1. Introduction ...
16
3.2 Results...17
3.2.1 Construction of combinatorial gRNA library ... 17
3.2.2 Efficiency of combinatorial gRNA expression system ... 19
3.2.3 Construction of combinatorial epigenetic perturbation library ... 19
3.2.4 Massively parallel combinatorial perturbation of epigenetic genes in cancer... 22
3.2.5 Identification of epigenetic gene combinations inhibit cancer cell growth... 25
3.3 Discussion ...
28
3.4 M ethods and M aterials ...
30
3.4.1 Vector Construction... 30
3.4.2 Assembly of the barcoded combinatorial sgRNA library pool ... 31
4 ... .... - - , ,%I,-- " 1-1, -- ,--,4
3 .4 .3 C ell culture ... 33
3.4.4 Lentivirus production and transduction ... 33
3.4.5 Sample preparation for barcode sequencing... 34
3.4.6 Barcode sequencing data analysis ... 35
3.4.7 Cell viability assay ... 36
3.4.8 Drug synergy quantification ... 37
3.4.9 Flow cytometry ... 37
3.4.10 Fluorescence microscopy ... :... 38
3.4.11 Quantitative PCR (qRT-PCR) ... 38
3.4.12 Immunoblot analysis ... 38
3.4.13 Surveyor assay ... 38
3.4.14 Sequencing analysis for indel detection ... 39
3.4.15 RNA-Seq and data analysis ... 42
3.4.16 M athematical modeling of cell proliferation in a mixed population ... 42
3.5 Supporting Information... 45
4. Genomic based memory recording in mammalian cells ...
70
4.1 Synthetic cellular memory circuits ... 70
4.1.1 Recombinased based genomically encoded cellular memory ... 70
4.1.2 Transcriptional regulation based cellular memory ... 72
4.1.3 Alternative molecular mechanism to build cellular memory ... 72
4.2 Genomically encoded analogy memory to record molecular signals...73
4.3 Continuous genetic recording with Self-targeting CRISPR-Cas in human cells...74
4.3.1 Modifying an sgRNA-expressing DNA locus to include a PAM renders it self targeting .76 4.3.2 stgRNA-encoding loci undergo multiple rounds of self-targeted mutagenesis...80
4.3.4 Small-molecule inducible and multiplexed memory storage using mSCRIBE ... 91
4.3.5 Recording the activation of the NF-kB pathway via mSCRIBE ... 91
4.3.6 Recording LPS-inducible inflammation in vivo via mSCRIBE... 92
4.4 M ethod and material ...
96
4.4.1 Vector construction ... 96
4.4.2 T7 Endonuclease I (T7 El) assay and Sanger sequencing ... 96
4.4.3 Cell culture, transfections and lentiviral infections ... 97
4.4.4 Clonal cell lines and DNA constructs ... 97
4.4.5 Design of longer stgRNAs... 97
4.4.6 FACS and microscopy ... 98
4.4.7 Mutation-Based Toggling Reporter (MBTR)-based cell sorting experiment ... 98
4.4.8 Next-generation sequencing and alignment ... 99
4.4.9 Barcoded stgRNA sequence evolution and transition probabilities ... 99
4.4.10 Small-molecule-inducible and multiplexed memory storage ... 101
4.4.11 In vivo inflammation model ... 101
4.5 Supporting information ... 103
4.6 Discussions...119
5. Future outlook and alternative approaches to build memory recorders...120
5.1 Epigenetics based memory recorder...121
5.2 Alternative approaches to build memory recorder ... 122
5.2 Future direction and application of memory recorder system ... 122
6. References...124
7. Appendix...136
List of Figures
Fig. 3-1. Strategy for Assembling Barcoded Combinatorial gRNA Libraries... 18 Fig. 3-2. High-Throughput Screen Identifies gRNA Combinations that Inhibit Cancer Cell Proliferation.
... 2 4 Fig. 3-3. Combinatorial Inhibition of KDM4C and BRD4, as well as KDM6B and BRD4, Inhibits Human
O varian C ancer C ell G row th. ... 27 Fig. 3-S1. Strategy for Assembling the Barcoded gRNA Library Pool... 45
Fig. 3-S2. Lentiviral Delivery of Combinatorial gRNA Expression Constructs Provides Efficient Target G ene R epression . ... 47 Fig. 3-S3. Generation of a High-Coverage Combinatorial gRNA Library and Efficient Delivery of the
L ibrary to H um an C ells. ... 49 Fig. S4. Activity of gRNAs at Targeted Genomic Loci in OVCAR8-ADR-Cas9 Cells...51 Fig. 3-S5. Deep Sequencing for Indel Analysis at gRNA-Targeted Genomic Loci in OVCAR8-ADR-Cas9
c e lls . ... 5 3
Fig. 3-S6. Sequencing of Targeted Alleles for Individual OVCAR8-ADR-Cas9 Cells Infected with Dual-gR N A E xpression C onstructs...56 Fig. 3-S7. Mathematical Modeling of the Frequency of a Pro-Proliferative gRNA and an
Anti-Proliferative gRNA within a Mixed Cell Population... 58 Fig. 3-S8. Fold-Changes in Barcodes among the Same gRNA Combinations Arranged in Different Orders
in the E xpression C onstructs...59 Fig. 3-S9. Biological Replicates and Log2 Fold-Change Comparisons for the Combinatorial Screen Used
to Identify gRNA Pairs that Inhibit Cancer Cell Proliferation. ... 60 Fig. 3-S 10. Pooled Screen and Validation Data for Individual gRNA Combinations... 62 Fig. 3-S 11. Measurement of On-Target and Off-Target Indel Generation Rates for gRNAs Targeting
Fig. 3-S 12. Reduced Growth in OVCAR8-ADR-Cas9 Cells Harboring Both KDM4C and BRD4
Fram eshift M utations... 64
Fig. 3-S 13. shRNA-Mediated Knockdown of Targeted Genes in OVCAR8-ADR Cells... 65
Fig. 3-S14. RNA-Sequencing Analysis of OVCAR8-ADR-Cas9 Cells Infected with gRNA Expression C onstructs. ... 66
Fig. 3-S 15. Effect of KDM4C and BRD4, as well as KDM6B and BRD4, on Cell Growth for Additional C ancer C ell L ines... 68
Fig. 4-1 Continuously evolving self-targeting guide RNAs. ... 79
Fig. 4-2 Tracking repetitive and continuous self-targeting activity at the stgRNA locus. ... 83
Fig. 4-3 stgRNA sequence evolution analysis. ... 90
Fig. 4-4 mSCRIBE as an analog memory device in vitro and in vivo ... 94
Fig. 4-SI Sanger sequencing of stgRNA loci confirming self-targeting CRISPR-Cas activity... 103
Fig. 4-S2 Validating functionality of the Mutation-Based Toggling Reporter (MBTR) system with different indel sizes in the Mutation Detection Region (MDR). ... 104
Fig. 4-S3 Sanger sequencing of stgRNA loci from sorted cells containing the self-targeting Mutation-Based Toggling Reporter (MBTR) construct. ... 105
Fig. 4-S4 Computational analysis of stgRNA sequence evolution from the barcoded stgRNA library experim ent... 107
Fig. 4-S5 Analysis of stgRNA sequence variants in the 'MIXD' format. ... 109
Fig. 4-S6 Transition probability matrix for 30nt-1 stgRNA . ... 112
Fig. 4-S7 Regular sgRNAs as memory operators ... 113
Fig. 4-S8 Small-molecule inducible mSCRIBE memory operators ... 115
Fig. 4-S9 Characterization of in vitro (A-C) and in vivo (D-E) NF-kB-responsive gene expression to TN Fa stim ulation and LPS, respectively. ... 116
Fig. 4-S 10 Percent mutated stgRNA metric measured for different number of cells... 118
Table S i. sgRNA Target Sequences Used in this Study... 136
Table S2. List of PCR Primers Used in qRT-PCR ... 140 Table S3. Sequencing of Targeted Alleles in OVCAR8-ADR-Cas9 Single Cells Harboring BMI1-sg2 and
PH F8-sg2 Expression C onstruct ... 143 Table S4. List of Two-Wise gRNA Hits that Inhibit OVCAR8-ADR Cell Proliferation Based on Pooled
S creen in g ... 14 4 Table S5. Constructs U sed in This W ork... 146 Table S6. shRNA Antisense Sequences Used for Individual Validation Assay ... 148 Table S7. Barcode Counts for the Pooled Screen ... 149 Dataset S8. Log2 Ratios Determined for Two-Wise gRNA Combinations Based on Pooled Screening .150
Table S9. List of PCR Primers Used in Surveyor Assay and Sanger Sequencing for Genome Modification D etectio n ... 15 1 Table S10. List of PCR Primers Used in Deep Sequencing for Indel Detection ... 152
1. General Introduction
The complex, dynamic, and responsive behavior of cells arises from integrated signaling pathways and regulatory networks. Previous efforts in building genetic circuitry to recapitulate and regulate biological behavior have been mainly focused on programming microbes due to their ease of genetic manipulation, rapid reproduction rate and well-characterized regulatory elements. With recent advancements in our ability to engineer mammalian cells, we concentrated our effort towards advancing mammalian synthetic biology. We aim to harness the rapidly evolving molecular tools to develop synthetic biology-enabled applications in mammalian cells. In this work, we designed and built genetic circuits and network systems to help answering biological questions in health and in diseases.
The RNA guided DNA endonuclease Cas9 from the Clustered Regularly Interspaced Short Palindromic Repeats(CRISPR)-associated (Cas) prokaryotic adaptive immune system is the newest tool in the mammalian cell genome engineering toolkit. The Cas9 nuclease enables sequence specific recognition guided by the complexing guide-RNA(gRNA). The high efficiency and versatility of the CRISPR-Cas system led to its widespread adoption. In my thesis work, I utilized the CRISPR-Cas system to construct a combinatorial genetic perturbation platform and re-engineered the system to encode longitudinal analog memory in the genome.
This thesis work consists of two major projects. First, we built a massively parallel genetic perturbation platform to decode the combinatorial genetics governing the complex regulation of cellular behavior. The system allows systematic and efficient interrogation of multiple genetic components in high-throughput and facilitates the understanding of gene networks. The second part of the work consists of the establishment of an analog cellular memory system encoded in genome by re-purpose the CRISPR-Cas9 system to enable self-targeting. The new cellular memory system allows long-term recording and storage of molecular events in the cell's genome, which can be later retrieved via next generation sequencing.
10
2. Massively parallel combinatorial genetic perturbation screening
2.1 Combinatorial genetics in modern biology
The complex networks of genetic regulators encoded in the genome control biological processes. Multifactorial and complex regulatory circuits are integral to a wide range of important biological phenomena relevant to human health and disease. Regulatory circuits in cells process signals, make appropriate decisions, and orchestrate physiological responses under diverse signals. Diseases, in turn, arise from circuit malfunctions: one or more components are missing or defective; key modules are over-or under-active. To understand complex diseases and develop mover-ore effective treatments, we need a comprehensive picture of all the cellular components, the circuits in which they function, and how these integrate to form physiological and malfunctioning responses.
With previous efforts in the Human Genome Project and the ENCODE (Encyclopedia of DNA elements) Project, we now have a more comprehensive annotation of functional genetic elements in human genome, which have paved the path for inquiring their functions (1). Advances in high-throughput assays have enabled massively parallel functional characterization of individual genetic elements on a genome-wide scale (2-6). Genomic research on dissecting cellular circuitry has progressed from genomic observations that enable statistical correlations between regulators and their targets to perturbation of single components at genome-wide scale. Evidently, compensatory mechanisms and cooperative actions between genetic regulators, as well as gene redundancies, contribute to the multilayered regulation in the genetic network functions (7-10). Testing genes individually is too limited: because the genes in biological circuits have non-linear interactions, we cannot predict how a circuit functions simply by summing up the individual effects. This is a direct outcome of the biochemistry underlying molecular biology, from allosteric protein changes to cooperative binding, and is essential for cells to process complex signals. The network connectivity has been so far determined based on experimentally perturbed individual- or paired- gene functions, which could fail in capturing systems-level effects due to higher-order interactions of the genetic regulators. It has remained an insurmountable stumbling block to
achieving a quantitative and predictive understanding of circuits on a genomic scale, with far-reaching implications for basic and translational science.
2.2 Current approaches in studying combinatorial genetics
Systematic experimental discovery of combinatorial effectors is limited by traditional methods, which are costly, time consuming, labor intensive, and not scalable to high diversity or combinatorial complexity. Rationally-designed and systematic implementation of combinatorial screening platform is necessary towards understanding of complex biological system. Diverse genetic perturbation tools and high-throughput assays have been developed for functionally characterizing the genetic elements via large-scale genetic screens, such as overexpression construct, RNA interference technology, and gene editing tools. Development in technology platforms enabled massively parallel studies to further increase the throughput and complexity of the screening. With discovery of a diverse set of perturbation tools and technologies for high-thorughput combinatorial genetics profiling analysis, comprehensive studies to decipher complex genetic circuitry became possible. The ability to generate gene perturbations has been pivotal to understand gene functions. Gene expression constructs, microRNA expression constructs, RNA interference (RNAi)-mediated knockdown of gene and long non-coding RNA (IncRNA), and gene-engineering tools like clustered regularly-interspaced short palindromic repeats (CRISPR)-based gene knockout/repression/activation systems have been adopted in genetic screens and made feasible on a genome-wide scale, which will be described in later section.
Current methods conducting combinatorial screening are limited in throughput and complexity. Single genetic element overexpression libraries can be screened for cellular phenotypes but lack combinatorial complexity. Individually constructing and testing multi-gene element constructs is laborious and expensive due to the large number of possible combinations. Pooled PCR stitching (11) and pairwise assembly (12) methods permit higher-throughput screening of pairwise gene perturbations but do not scale easily to combinations of 3 or more elements. The expense and duration of such studies impede
12
the discovery of novel combinatorial therapies. Thus, there is a pressing need for systematic, high-throughput, and scalable technologies to discover novel combinatorial therapeutics.
2.3 Genome engineering tools as a gene perturbation platform
Genome-engineering tools offer the next generation platform for gene perturbation. The advent of the genome-engineering tools, including artificial zinc finger (ZF)-, transcription activator-like effector (TALE)-, and CRISPR- based systems, provides us the opportunities to generate genetic perturbations in a programmable manner. The programmability and efficiency of the CIRPSR-based system led to the widespread adoption of the technology as a gene perturbation platform.
In the CRISPR-based system, a customized short guide RNA (gRNA) directs the Cas9 nuclease to bind on the target genomic DNA site harboring the protospacer adjacent motif (PAM) sequence, create double-strand breaks, and then generate frameshift mutations. Thus, by simply substituting with another gRNA, the same Cas9 nuclease can be used to target different genomic DNA regions. Guide RNA can be readily generated by oligo synthesis technology, which thus circumvents the need to engineer and assemble the nuclease for individual targets. The discovery and development of these engineered nucleases (13, 14), as well as the practical guides (15), were reviewed in detail previously. In particular, the Cas9 nuclease derived from S. pyogenes (SpCas9) has been widely used and extensively characterized
(16, 17). To enhance robustness of the CRISPR-based system to generate gene knockouts, computational algorithms and machine learning-based predictive modeling have been developed to design gRNAs with maximal on-target and minimal off-target activities, which was demonstrated effective for SpCas9 (18-20). Such efforts led to the creation of several genome-wide gRNA libraries that can be used for studying gene knockout functions in a range of in vitro and in vivo models. Additional CRISPR-based nucleases, including Cpfl (21) and the Cas9 nucleases of other bacterial species such as S. aureus (SaCas9) and S.
thermophilius (StlCas9) (22-24), were identified to provide smaller-sized Cas9 nucleases for
genome-engineering applications and also the possibility for orthogonal gene-targeting via their corresponding PAM sequences. The protein sequence of Cas9 was also rationally engineered based on its structure
prediction and random mutagenesis to further reduce its off-target effect (25, 26) and alter its PAM specificities for a broader targeting spectrum (23, 27). In addition to knocking out genes, the Cas9 nucleases have further been applied for large-scale characterization of gene regulatory regions like the enhancer regions (28), as well as genetic mutations when coupled with homology directed repair strategies for saturation editing of the targeted genomic region (29).
The genome-engineering tools are highly versatile and have been repurposed for gene repression and activation studies. The fusion of the Krilppel associated box (KRAB) repression domain to the ZF and TALE proteins allows the recruitment of the domain to the specific site in the gene promoter region to inhibit transcription (30-33). A nuclease-dead version of Cas9, termed deactivated Cas9 (dCas9), with or without its fusion to the KRAB domain (dCas9-KRAB), can be expressed with a gRNA that targets the proximal region of the transcription start site in the genome to effectively repress gene transcription (34, 35). Because of its ease to design gRNAs that can be generated via oligo synthesis, gRNA libraries were created and applied for genome-wide gene repression screens (i.e., CRISPR interference (CRISPRi) screens)(36). The CRISPRi system was further shown to effectively inhibit lncRNA expression (36).
Using a similar strategy, the ZF, TALE, and dCas9 can be fused with the VP16-based activation domain (AD) to turn on gene transcription (34, 35, 37-43). It was realized, however, that the fusion of the single VP16-based AD is often not sufficient to drive a high level of targeted gene activation, and multiple ZF-ADs, TALE-ZF-ADs, or gRNAs for the dCas9-AD, that target regions near the transcription start site in concert could enhance the extent of transcriptional activation (38-43). However, the rules for predicting which combinations of the ZF-ADs, TALE-ADs and gRNAs, to use remain unknown and the rational design for gene activation could be difficult to achieve. Improved CRISPR-based gene activation systems including the CRISPR activation (CRISPRa)(36), the CRISPR-Synergistic Activation Mediator (SAM)(44), and the CRISPR-VP64-p65-Rta (VPR)(45) systems were then developed to allow single gRNA to be used for efficient gene activation. With these systems, more units of activation domains are recruited to the target genomic loci to mediate robust gene activation. Genome-scale gRNA libraries were
further created for the CRISPRa (36) and CRISPR-SAM (44) systems to query gene activation functions in mammalian cells. Multiplexed genetic perturbations can be effectively achieved using the CRISPR-based genome engineering tools. For example, CRISPR (46) and CRISPR-SAM (44) systems were shown to successfully knock out five and activate ten genes simultaneously, respectively.
3. Massively parallel combinatorial genetic perturbation screening with
CRISPR-Cas9 in human cells
This work is completed with Dr. Alan Wong and Dr. Gigi Choi as equal contributors, and this entire
chapter has been published as in PNAS 113.9 (2016): 2544-2549.
3.1. Introduction
New therapeutic strategies are needed to treat complex human diseases. Since disease phenotypes are often regulated by interwoven genetic networks, exploiting combination therapy to target multiple pathways, as opposed to only single ones, can enhance treatment efficacy (47). However, discovering effective combination therapies for human diseases is challenging with existing methods due to the cost, effort, and labor required to construct and analyze each combination (48). For example, the National Cancer Institute tested -5,000 pairwise combinations of 100 cancer drugs against the NCI-60 panel in a study that took two years and cost about USD$4 million (49). Thus, there is a need for technological advances to accelerate the identification of effective combinatorial therapies. Here, we used our CombiGEM-CRISPR platform to perform rapid pooled screening of pairwise genetic knockouts against genes coding for epigenetic regulators and then translated our screen hits into novel drug combinations against human ovarian cancer cells.
CRISPR-Cas9 technology has been used for large-scale genetic perturbation screens with single guide RNA (sgRNA) libraries for gene knockouts (50-53), repression and activation (36, 54). Despite its simplicity for multiplexed genetic perturbations (34, 55, 56), new methods are needed to enable high-throughput CRISPR-Cas9-based screening with combinatorial sets of gRNAs, which would be broadly useful for studying combinatorial gene functions in multigenic phenotypes and diseases. By using CombiGEM-based DNA assembly (57, 58), we developed a strategy for the simple and efficient assembly of barcoded combinatorial gRNA libraries. These libraries can be delivered into human cells by lentiviruses in order to create genetically ultra-diverse cell populations harboring unique gRNA combinations that can be tracked via barcode sequencing in pooled assays. This strategy, termed
CombiGEM-CRISPR, uses one-pot cloning steps to enable the assembly of combinatorial gRNA libraries, thus simplifying and accelerating the workflow towards systematic analysis of combinatorial gene functions.
3.2 Results
3.2.1 Construction of combinatorial gRNA library
To create the initial barcoded sgRNA library, an array of oligo pairs encoding a library of barcoded gRNA target sequences was first synthesized, annealed, and pooled in equal ratios for cloning downstream of a U6 promoter in the storage vector (Fig. 3-1; Fig. 3-Si). Subsequently, the scaffold sequence for the gRNAs was inserted into the storage vector library in a single-pot ligation reaction. We then applied the CombiGEM method for assembly of combinatorial gRNA libraries (Fig. 3-1). Within the barcoded sgRNA construct, BamHI and EcoRI sites were positioned in between the gRNA sequence and its barcode, while BglII and MfeI sites were located at the ends. Strategic positioning of these restriction enzyme sites resulted in the segregation of the barcode from its gRNA sequence upon enzymatic digestion and the concatenation of barcodes representing their respective gRNAs upon ligation of inserts. To construct the one-wise library, pooled inserts of the barcoded sgRNA expression units were prepared by restriction digestion of the storage vectors with BglII and MfeI and joined to their compatible DNA ends in the lentiviral destination vector, which was digested with BamHI and EcoRI. The one-wise library then served as the destination vector for the next round of pooled insertion of the barcoded sgRNA expression units to generate the two-wise library, in which barcodes representing each sgRNA were localized to one end of each lentiviral construct. This process can be iteratively repeated to generate higher-order barcoded combinatorial gRNA libraries. The identity of the combinatorial gRNAs can be tracked by high-throughput sequencing of the concatenated barcodes, which are unique for each combination.
1. Oligo synthesis 2. Annealing of complementary 3. Pooled insertion of gRNA of barcoded gRNA oligos for pooled library cloning scaffold sequence
target sequences
gRNA target gRNA target
sequence sequence
'I / scffold
U6p U6p . sca Barcode
Bbs Bgll BamHl EcoRi Mfel
Oligo array Storage vector library Barcoded sgRNA library
4. (n)-round pooled ligation of barcoded inserts digested with Bglll + Mfel and vectors digested with BamHl + EcoR
BamHl EcoRI _______________
Lentiviral vector----
---backbone
U6p
BamHl EcoRI 1-wise gRNA sgRNA-A
library- l~brary __---__--- -. ..- . ..- - - -
-U6p U6p
BamHl EcoRi
2-wIse gRNA sgRNA-A sgRNA-B
library ---.4
(n) x sgRNA (n) x barcodes
~,~.g~
g W..2a rnrco l I~ o(n)-wlse gRNA
library II'
Concatenated CombiGEM barcodes representing gRNA combinations
Fig. 3-1. Strategy for Assembling Barcoded Combinatorial gRNA Libraries.
Barcoded gRNA oligo pairs were synthesized, annealed, and cloned in storage vectors in pooled format. Oligos with the gRNA scaffold sequence were inserted into the pooled storage vector library to create the barcoded sgRNA library. Detailed assembly steps are described in Fig. 3-Si. The CombiGEM strategy was used to build the combinatorial gRNA library. Pooled barcoded sgRNA inserts prepared from the sgRNA library with BglII and MfeI digestion were ligated via compatible overhangs generated in the destination vectors with BamHI and EcoRI digestion. Iterative one-pot ligation created (n)-wise gRNA libraries with unique barcodes corresponding to the gRNAs concatenated at one end, thus enabling tracking of individual combinatorial members within pooled populations via next-generation sequencing.
3.2.2 Efficiency of combinatorial gRNA expression system
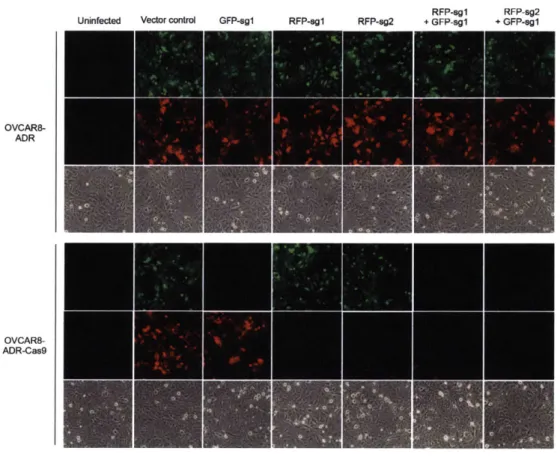
To evaluate the functionality of our lentiviral combinatorial gRNA expression system, we built gRNA combinations targeting green fluorescent protein (GFP) and red fluorescent protein (RFP) sequences (Appendix, Table Sl) and determined the combinatorial gene perturbation phenotypes using flow cytometry (Fig. 3-S2A-D) and fluorescence microscopy (Fig. 3-S2E). Lentiviruses carrying dual RFP and GFP reporters together with the barcoded combinatorial gRNA expression units were used to infect human ovarian cancer cells (OVCAR8-ADR) (59) stably expressing human codon-optimized Cas9 nuclease (OVCAR8-ADR-Cas9) (Fig. 3-S2A). We anticipated that active gRNAs would target the GFP and RFP sequences, and generate indels to knockout the expression of GFP and RFP. Efficient repression of GFP and RFP fluorescence levels was observed, as the GFP and RFP double-negative population was the major one observed in cells carrying both Cas9 nuclease and double gRNA expression units at both day 4 and 8 post-infection (-83 to 97% of the total population), compared with < 0.7% in the vector control (Fig. 3-S2C, D). This repression was not observed in control cell lines expressing the gRNAs targeting GFP and/or RFP but without Cas9 nuclease (Fig. 3-S2B). The specificity of gene perturbation was confirmed, as cells harboring GFP-targeting sgRNA exhibited loss of the GFP signal but not the RFP signal, and vice versa for cells with the RFP-targeting sgRNAs (Fig. 3-S2E). These results demonstrate the ability of lentiviral vectors to encode combinatorial gRNA constructs that can repress the expression of multiple genes simultaneously within a single human cell.
3.2.3 Construction of combinatorial epigenetic perturbation library
Using CombiGEM-CRISPR, we sought to discover combinatorial epigenetic perturbations with anti-cancer phenotypes, since diverse epigenetic modifications tend to act cooperatively to regulate gene expression patterns (60) and combinatorial epigenetic modulation is emerging as a promising strategy for cancer therapeutics (61, 62). We constructed a library of 153 barcoded sgRNAs targeting a set of 50 genes that encode epigenetic regulators (3 sgRNAs per gene) and 3 control sgRNAs based on the GeCKOv2 library (50) (Appendix, Table Si). Using the Drug-Gene Interaction database (DGIdb) (63)
and recent literature (64, 65), we confirmed that at least 26 out of the 50 genes in our library are known drug targets. Epigenetic protein families belong to druggable classes of enzymes or cofactors against which an increasing list of drugs are undergoing pre-clinical or clinical development (64).
We confirmed the expression of these 50 genes in OVCAR8-ADR cells using qRT-PCR (Appendix, Table S2). We then created a two-wise (153 x 153 sgRNAs = 23,409 total combinations) pooled barcoded gRNA library using CombiGEM. Lentiviral pools were produced to deliver the library into OVCAR8-ADR-Cas9 cells, and genomic DNA from the pooled cell populations was isolated for unbiased barcode amplification by polymerase chain reaction (PCR). Illumina HiSeq was used to quantify the representation of individual barcoded combinations in the plasmid pools stored in Escherichia coli and also in the infected human cell pools (Fig. 3-S3A-D). We achieved high coverage for the two-wise library within both the plasmid and infected cell pools from -23 to 34 million reads per sample (Fig.
3-S3B), and a relatively even distribution of barcoded gRNA combinations was observed (Fig. 3-S3A, B).
Furthermore, we observed highly correlated barcode representation between the plasmid and infected cell pools (Fig. 3-S3C), as well as high reproducibility in barcodes represented in biological replicates for infected cell pools (Fig. 3-S3D). Thus, CombiGEM-CRISPR can be used to efficiently assemble and deliver barcoded combinatorial gRNA libraries into human cells.
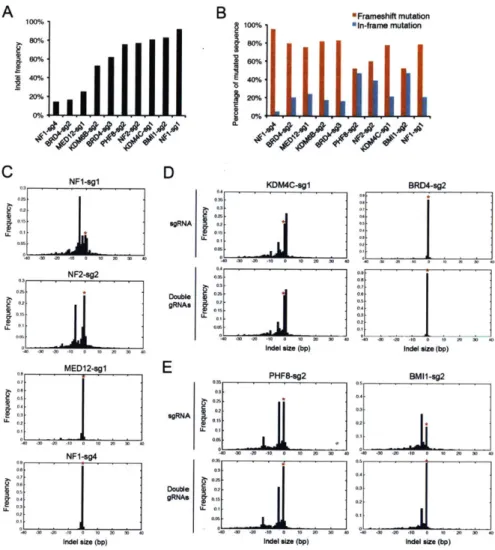
Previous CRISPR-Cas9-based gene knockout screens have demonstrated high genomic modification efficiencies after about 6 to 12 days post-expression of Cas9 and sgRNA in human cells. However, it is important to evaluate genomic modification efficiencies in the specific cell types to be studied due to variations among gRNAs and cell types (50-52). To confirm the ability of the CRISPR-Cas system to edit endogenous genes in OVCAR8-ADR-CRISPR-Cas9 cells, we performed Surveyor assays to detect mutations at genomic loci targeted by 8 randomly chosen gRNAs from our library. We observed cleavage of DNA mismatches for all of the gRNA-targeted loci at day 12 post-infection (Fig. 3-S4A-C). We further determined the simultaneous cleavage efficiency at multiple loci in our dual-gRNA system, and observed comparable levels of cleavage in cells expressing individual gRNAs or double gRNAs (Fig.
3-S4A, D). Depletion of targeted protein levels in individual gRNA- and double gRNA-expressing cells
was also detected (Fig. 3-S4E). These results suggest that our multiplexed system does not hamper the activity of gRNAs.
We next estimated indel generation efficiency by performing deep sequencing at targeted genomic loci. Consistent with previous reports (50-52), we observed large variations in the rates of generating indels (i.e., 14 to 93%; Fig. 3-S5A) and frameshift mutations (i.e., 52 to 95% out of all indels; Fig. 3-S5B) among different gRNAs. In addition, gRNAs that were validated in a previous study with A375 melanoma cells (50) displayed reduced activity (e.g., for NF1-sg4 and MED12-sgl sgRNAs) and differential indel generation preferences (e.g., for the NF1-sgl sgRNA) in OVCAR8-ADR-Cas9 cells (Fig. 3-S5C). Such discrepancies could be partially due to variations in chromatin accessibility at target loci (66) and DNA break repair mechanisms (67) that can vary among cell types. Continual efforts in gRNA design optimization, including improving on-target cleavage rates (68)and minimizing off-target cleavage, should enable the creation of more efficient gRNA sets that will improve their applicability for large-scale genetic perturbation screening in a broad range of cell types. We further assessed indel generation by gRNAs in our multiplexed system. Our deep sequencing analysis detected largely comparable indel generation frequencies and preferences for the same gRNA expressed under the sgRNA or double gRNA systems (Fig. 3-S5D). To distinguish dual-cleavage events directed by double gRNAs within a single cell from cleavage events distributed across the population, we isolated clones derived from single cells infected with double gRNA constructs and were able to detect cells with insertions, deletions, or mutations in both targeted genomic loci (Fig. 3-S6A-C; Appendix, Table S3). Our results indicate that our combinatorial gRNA library can be used to generate double genetic mutants in OVCAR8-ADR-Cas9 cells. However, we believe that improvements in the efficiency of CRISPR-Cas reagents for gene knockouts would yield higher-quality CombiGEM-CRISPR libraries.
We initiated a pooled combinatorial genetic screen with OVCAR8-ADR-Cas9 cells to identify gRNA combinations that regulate cancer cell proliferation. We constructed a mathematical model to map
out how relative changes in abundances of each library member within a population depend on various parameters (see Section 3.4 Methods and Material; Fig. 3-S7A, B). We simulated populations containing heterogeneous subpopulations that harbor different gRNA combinations. Specifically, we defined specific percentages of the overall population at the start of the simulation as harboring subpopulations with anti-proliferative (f,) and pro-anti-proliferative
(ff)
gRNA combinations. Within each subpopulation, a fraction of cells was mutated by the CRISPR-Cas9 system (p) at the start of the simulation, resulting in a modified doubling time (Tdoubling,m). Our model indicated that the representation of barcoded cells with an anti-proliferative gRNA set in the entire cell population can be depleted by about 23 to 97% under simulated conditions (i.e.,fs,
and ff = 2, 5, or 10%; p = 0.2, 0.4, 0.6, 0.8, or 1.0; Tdoublingm= 36, 48, or 60 hours) (Fig. 3-S7B). In general, increasing mutation efficiencies, increasing doubling times for anti-proliferative cells, decreasing doubling times for proliferative cells, as well as increasing the percentage of pro-proliferative combinations in the population (Fig. 3-S7C), are expected to result in greater barcode depletion of anti-proliferative barcodes in the overall population.3.2.4 Massively parallel combinatorial perturbation of epigenetic genes in cancer
In the experimental screen, we cultured OVCAR8-ADR-Cas9 cell populations infected with the two-wise combinatorial gRNA library for 15 and 20 days, and isolated their genomic DNA for unbiased amplification and quantification of the integrated barcodes (Fig. 3-2A; Fig. 3-S3E, F). Since phenotypes resulting from progressive epigenetic alterations following targeted gene inactivation are expected to take time to manifest (69), barcode abundances (normalized per million reads) between the day 15 and day 20 groups were compared to yield log2 (barcode count ratios) values (Fig. 3-2A; Fig. 3-S3E, F), based on similar time windows used in previous studies on the anti-proliferative effects resulting from epigenetic perturbations (69, 70). To reduce variability, combinations with less than -100 absolute reads in the day 15 group were filtered out, and the log2 ratios of the two possible arrangements for each gRNA pair (i.e.,
sgRNA-A + sgRNA-B and sgRNA-B + sgRNA-A) were averaged (Fig. 3-S8). Less than 4.4% of all
combinations were detected at <100 absolute reads in the day 15 group in both sets of experiments. The
correlation between the log2 ratios of the two possible arrangements for each gRNA pair could be improved by increasing the fold representation of cells per combination in the pooled screen to reduce experimental noise, as previously noted in pairwise genetic perturbation screens (71). Log2 ratios for each
gRNA combination were determined for two biological replicates and ranked (Fig. 3-2B; Fig. 3-S9A). The majority of the gRNA combinations did not exhibit significant changes in barcode representations between the day 15 and day 20 groups, including three control gRNAs from the GeCKOv2 library (50) that do not have on-target loci in the human genome as internal controls. We defined 61 gRNA combinations as top hits that exerted considerable anti-proliferative effects (log2 ratio < -0.90) in both biological replicates (Q-value <0.01; Fig. 3-S9B, C; Appendix Table S4), yielding potential sets of genes to investigate further for their ability to suppress the growth of cancer cells. A potential caveat to consider when comparing barcode abundances between the day 15 and day 20 groups is that cells with strongly synthetic-lethal gene combinations inactivated could be eliminated before day 15, leading to false negatives. To account for this, we compared log2 ratios between day 15 and day 5 groups, and did not identify any hits (Fig. 3-S9D). We speculate that this could be due to latency in proliferation changes resulting from the time required to alter epigenetic marks and gene expression after knockouts occur (69,
70). This analysis highlights the need to identify optimal time windows for performing barcode
comparisons to facilitate hit identification in high-throughput pooled screens.
CRISPR-Cas9-based screens, like other genetic screens, can lead to false discovery due to off-target effects and false-positive hits (3, 4). Thus, we performed individual validation experiments to verify the phenotype-modifying effects of genetic perturbations identified as hits from our screen. We validated screen hits by demonstrating their ability to inhibit the proliferation of OVCAR8-ADR-Cas9 cells in individual (non-pooled) cell growth assays using the corresponding gRNA pairs delivered via lentiviruses (Fig. 3-2C; Fig.3-S1O). In addition, false-negative hits could be anticipated in the screen (4,
72) due to the presence of inefficient gRNAs (Fig. 3-S5; Appendix Table Si), which could be addressed with an experimentally validated set of gRNAs with optimized on-target efficiencies.
A
agRNA-A sgRNA-B
Cell proltwatlton screew
2-wise banoded gRNA library
-- es Dap2it
OVCAR8-ADR-Caeg cells Dy2
Replcae 1 3.0 2.0- Vn1.0-0.0 -1 .0 -.0 -3.0 -4.0 -5.01 -6.0 Ranked barcoded -. cornbinations
-9gRNA-A + control sgRNA
BRD4-ag3 + PHF2.egl aAD4-ag3 +PHF24.-g i +R" * -W202 Ranked barcoded combinations -9gRNA-A + SgRNA-B 0.35 0.30 -- .vector controt 0.25 -0.25 -v- BRO&3+ KDM4.-M2 --- BR04-g3.+ KDM6B-692 0.20 -- BRD4-ag3 + PHF24gi .o BRD44g3 + PHF2-og2 0.15 0.10 0.05 0.00 1 2 3 4 5 Day
Fig. 3-2. High-Throughput Screen Identifies gRNA Combinations that Inhibit Cancer Cell Proliferation.
(A) OVCAR8-ADR-Cas9 cells infected with the barcoded two-wise gRNA library were cultured for 15
and 20 days. Barcode representations within the cell pools were quantified using Illumina HiSeq. (B) Two-wise gRNA combinations that modulated proliferation were ranked by log2 ratios between their normalized barcode counts in 20-day versus 15-day cultured cells (right panel). To enable comparisons between the two-wise gRNA combinations with their sgRNA counterparts, the same data for sgRNAs paired with control sgRNAs (left panel) is also plotted. Combinations with control gRNA pairs are highlighted in orange. The anti-proliferative effects of gRNA combinations that were confirmed in another biological replicate are highlighted in blue (see Fig. 3-S9). The labeled gRNA combinations were further validated in this study. (C) Individual validation of two-wise combinations that modulated cancer cell growth. OVCAR8-ADR-Cas9 cell populations individually infected with lentiviruses expressing the indicated two-wise gRNA combinations were cultured for 15 days, and equal number of cells were then
24
B
0U=~Idm
Barcodes Baooden 3.0- 2.0- 1.0- 0.0--1.0 -2.0- -3.0- -4.0- -5.0--6.0re-plated and cultured for additional time periods as indicated. Cell viability was measured by the MTT assay, and characterized by absorbance measurements (OD5 70 - OD6 5 0) (n = 3). Data represent mean SD.
3.2.5 Identification of epigenetic gene combinations inhibit cancer cell growth
Global alterations of epigenetic landscapes observed in cancer progression (73) and the reversible nature of epigenetic states (74) suggest that targeting multiple epigenetic regulators could help to suppress cancer growth. Interestingly, we observed that many gRNAs targeting genes encoding epigenetic regulators exhibited stronger anti-proliferative effects when used in combination with other gene-targeting gRNAs than when they were used in combination with control gRNAs (Fig. 3-2B; Fig. 3-S9A). With individual validation assays, we confirmed that specific gRNA pairs (Fig. 3-3A; Fig. 3-S4) targeting KDM4C and BRD4 simultaneously led to synergistic reductions in cancer cell growth. We also assessed the off-target activity of these gRNAs with deep sequencing, which revealed a low indel generation rate (i.e., 0.15 to 0.38%) at the exonic off-target genomic loci computationally predicted by the CRISPR design (75) and CCTop (76) tools for the two gRNAs (Fig. 3-S 11). In addition, we observed reduced growth in a single-cell-derived clone harboring both KDM4C and BRD4 frameshift mutations (Fig. 3-S12). Collectively, we established and validated an experimental pipeline for the systematic screening of barcoded combinatorial gRNAs that are capable of exerting anti-proliferative effects on ovarian cancer cells.
We further used RNA interference and small-molecule drugs to modulate genes encoding epigenetic regulators and confirm the screening-based phenotypes. Expression of multiple shRNA pairs targeting KDM4C and BRD4 (Fig. 3-3B; Fig. 3-S 13) and co-treatment with the small-molecule KDM4C inhibitor SD70 (77) and small-molecule BRD4 inhibitor JQ1 (78) (Fig. 3-3C) inhibited the proliferation of OVCAR8-ADR cells synergistically. Similarly, gRNA pairs (Fig. 3-3A; Figs. 3-S4 and 3-S11) and shRNA pairs (Fig. 3-3B; Fig. 3-S13) that simultaneously targeted KDM6B and BRD4 exhibited synergy, as did co-treatment with the small-molecule KDM6B/6A inhibitor GSK-J4 (79) and JQ1 (Fig. 3-3D). Synergy between both of these pairwise combinations of small-molecule drugs was confirmed by both the
Bliss independence (80) and the Highest Single Agent (81) models (Fig. 3-3C, D). These multiple
confirmatory strategies suggest that the observed anti-proliferation effects were likely caused by the dual-inhibition of multiple genes rather than off-target effects. Our approach thus facilitates the identification of novel interacting gene pairs that inhibit cancer cell proliferation, and the potential development of synergistic drug therapies.
B
t-1.2-4 1.1 - I * 9 1.0 I I T- 0.9-KDM4C-sgl KDM6B-sg2 BRD4-sg3 - + -- - + - + -- -4+ + + +C
Excess over Bliss Excess over
% independence model HSAmodsl
SD70 (pM) SD70 (pM) SD70 (pM) 0 1.25 2.5 0 1.25 2.5 C 0 0 0 CY 0.1 8 1 61 0 -5]e 0.2 4 8 0 1 1 * 50- F 40-30- Expected effect by Bliss e 0 independence 0 0 model 0 -10 JQ1 (0.2 pM) - - + + SD70 (1.25 pM) - + - + 1 .4-1 .2-1.0 0.8- 0.4-KDM4C-Shl KDM4C-sh2 KDMWB-eh BRD4-shl BRD4-sh2 Control-sh
D
A
% inh GSK-J4 0 1.25 =L 0 M2t6 0. _& 39t5 0.11 36 42* 60 0 2 0--20 JOI (0.8 pM) GSK-J4 (1.25 pM)i Excess over Bliss Excess over
independence model HSA model
(pM) GSK-J4 (pM) GSK-J4 (pM) 2.5 0 1.25 2.5 34t4 0 44 . 54 1 0 Expected
T
effect by Bi independence model 0 0 0 - - + + - + - +Fig. 3-3. Combinatorial Inhibition of KDM4C and BRD4, as well as KDM6B and BRD4, Inhibits
Human Ovarian Cancer Cell Growth.
(A and B) OVCAR8-ADR-Cas9 cells infected with lentiviruses expressing the indicated single or
combinatorial gRNAs (A) or OVCAR8-ADR cells co-infected with lentiviruses expressing the indicated shRNAs (B) were cultured for 15 days and 9 days, respectively. Equal numbers of infected cells were
0* - + - - - - + + - - -- - + --- ---- + + -- - - +--- - ---+ -- - - + - + - + - + - - ---+ -+ -+ -+ -iss
then re-plated and cultured for additional 5 days (A) and 4 days (B). Cell viabilities relative to control sgRNA (A) or shRNA (B) were determined by the MTT assay. (C and D) OVCAR8-ADR cells were treated with the indicated drugs for 5 days (C) and 7 days (D). JQ1 (78), SD70 (77), and GSK-J4 (79) are small-molecule inhibitors of BRD4, KDM4C, and KDM6B/6A, respectively. Percentage inhibition of cell growth relative to no drug control was determined by the MTT assay. The calculated excess inhibition over the predicted Bliss independence and HSA models was shown for each drug combination pair. Data represent mean SD (n =3 for (A); n = 6 for (B to D)) from biological replicates. The asterisk (*P < 0.05) and hash (#P < 0.05) represent significant differences between the indicated samples and between
drug-treated versus no drug control samples, respectively.
3.3 Discussion
In summary, we established CombiGEM-CRISPR as a technology platform for assembling barcoded combinatorial gRNA libraries and facilitating pooled genetic perturbation screening that can be translated into novel drug combinations. This platform expands the utility of CRISPR-Cas9-based systems for performing systematic multiplexed genetic perturbation screens in high-throughput. Here, we applied CombiGEM-CRISPR to perform a massively parallel combinatorial CRISPR-Cas screen and successfully isolated gene pairs for which simultaneous inhibition via CRISPR-Cas knockouts, RNA interference, and small molecules led to reduced ovarian cancer cell growth. High-throughput screening of combinatorial genetic perturbations by CombiGEM-CRISPR can expedite the identification of novel drug combinations with desired therapeutic effects by targeting libraries of gRNAs against druggable genes (63, 82). In this study, we further investigated two of our genetic hits (i.e., KDM4C + BRD4 and KDM6B + BRD4) with readily available small-molecule drugs and confirmed their synergistic efficacy against ovarian cancer cells. These drugs have been used previously for treating other cancer cell types in mouse models with limited toxicity (77, 78, 83, 84), suggesting that, in combination, these drugs
could be viable therapeutic candidates. Our approach advances upon previously described combinatorial
drug screening platforms (48, 81, 85, 86) by using multiplexed and pooled screens to reduce the cost, time, and effort required.
This strategy can also help identify new areas for biological inquiry, such as studies into the mechanisms that underlie observed phenotypes. For example, we analyzed gene expression patterns in cell populations infected with lentiviruses encoding gRNAs targeting both KDM4C and BRD4, or KDM6B and BRD4 (Fig. 3-S14A). Significantly perturbed genes were associated with gene sets involved in cancer-related pathways, including TNFa/NFiB signaling, p53 pathways, and apoptosis (Fig. 3-S14B). In addition, the combinatorial effects of epigenetic perturbations are complex and can vary across different cell types (Fig. 3-S15). In future work, assembled and barcoded combinatorial libraries could be directly delivered into a variety of cell types to rapidly dissect how combinatorial genetic effects vary based on genetic background via the same screening pipeline.
The utility of CombiGEM-CRISPR will be further enhanced as CRISPR-Cas technology continues to be improved in terms of enhanced cleavage efficiency (68, 82, 87) and reduced off-target effects (41, 88, 89). Variations in gRNA efficiency and indel generation preference can result in quantitative differences in creating disruptive mutations between gRNAs targeting the same gene. In addition, mixed genotypes generated by the CRISPR-Cas genome editing can generate variability in pooled screens. As new rules for designing highly efficient frameshift-mutation-creating gRNAs are established in the future (4), CombiGEM-CRISPR could be potentially applied to perform large-scale studies of epistasis for interrogating gene-gene interactions. Previous efforts have laid powerful experimental and computational foundations for performing systematic genetic interaction analysis in yeast and human cells (12, 90). In addition to generating gene knockouts, the CombiGEM-CRISPR platform could be used for high-order combinatorial gene activation and repression studies by incorporating gRNAs and deactivated Cas9 variants repurposed as transcriptional and epigenetic regulators (36, 54, 91, 92). This technology could also be used to interrogate the function of large genomic deletions (93) and rearrangements (94, 95) with barcoded gRNA pairs. Thus,
CombiGEM-CRISPR provides a facile approach to uncover gene and drug combinations that exert desired biological responses, especially for phenotypes that require more than a single perturbation to be manifested due to underlying complex biological networks.
3.4 Methods and Materials
3.4.1 Vector Construction
The vectors used in this study (Appendix Table S5) were constructed using standard molecular cloning techniques, including restriction enzyme digestion, ligation, PCR, and Gibson assembly. Custom oligonucleotides were purchased from Integrated DNA Technologies. The vector constructs were transformed into E. coli strain DH5a, and 50 pig/ml of carbenicillin (Teknova) was used to isolate colonies harboring the constructs. DNA was extracted and purified using Plasmid Mini or Midi Kits (Qiagen). Sequences of the vector constructs were verified with Genewiz's DNA sequencing service. The constructs for CombiGEM-CRISPR are available to the academic community through Addgene.
To generate a lentiviral vector encoding an shRNA that targeted a specific gene, oligo pairs harboring the sense and antisense sequences were synthesized, annealed, and cloned in the Agel- and EcoRI-digested pLKO.1 vector(96) (Addgene plasmid #10879) by ligation. The shRNA sense and antisense sequences were designed and constructed based on the siRNA Selection Program
(http://sima.wi.mit.edu/), and listed in Appendix Table S6.
To create the pAWp30 lentiviral expression vector encoding Cas9 protein and Zeocin as the selection marker, EFS promoter and Cas9 sequences were amplified from Addgene plasmid #49535, while the Zeocin sequence was amplified from Addgene plasmid #25736, by PCR using Phusion DNA polymerase (New England Biolabs). The PCR products were cloned into the pAWp 1 lentiviral vector backbone using Gibson Assembly Master Mix (New England Biolabs).
To construct a storage vector containing U6 promoter (U6p)-driven expression of a sgRNA that targeted a specific gene, oligo pairs with the 20 bp gRNA target sequences were synthesized, annealed, and cloned in the BbsI-digested pAWp28 vector using T4 ligase (New England Biolabs). To construct a lentiviral vector for U6p-driven expression of single or combinatorial gRNA(s), U6p-sgRNA expression cassettes were prepared from digestion of the storage vector with BglII and MfeI enzymes (Thermo Scientific), and inserted into the pAWpl2 vector backbone or the sgRNA expression vector, respectively, using ligation via the compatible sticky ends generated by digestion of the vector with BamHI and EcoRI enzymes (Thermo Scientific). To express the gRNAs together with the dual RFP and GFP fluorescent protein reporters, the U6p-driven sgRNA expression cassettes were inserted into the pAWp9, instead of pAWp12, lentiviral vector backbone using the same strategy described above. The pAWp9 vector was modified from the pAWp7 vector backbone by introducing unique BamHI and EcoRI sites into the vector to enable the insertion of the U6p-sgRNA expression cassettes.
3.4.2 Assembly of the barcoded combinatorial sgRNA library pool
We built a library of 153 barcoded sgRNAs targeting a set of 50 genes encoding epigenetic regulators (3 sgRNAs per gene) and 3 control sgRNAs based on the publicly available GeCKOv2 library for genome-wide gene-knockout screening (50) (Appendix Table Sl). We scanned through the GeCKOv2 library, and found that only a small number (-2.8%) of the gRNAs in the library contained the four restriction sites (i.e., EcoRI, MfeI, BamHI, and BglII) used for CombiGEM assembly. Among the 6 sgRNAs per gene in the GeCKOv2 library, less than 1.8% and 0.15% of genes have more than one and two of their sgRNAs, respectively, that contain any of these four restriction sites. In the library used in this study, only one sg$NA (i.e. PRMT3-sg3) contained an EcoRI recognition site. Our CombiGEM method is therefore compatible with existing gRNA libraries design for conducting large-scale screening experiments. Other restriction enzyme pairs that have compatible ends can also be used to assemble combinatorial gRNA expression vectors with CombiGEM.
An array of 153 oligo pairs (Oligo F-(x) and Oligo R-(x), where x = 1 to 153) harboring the barcoded gRNA sequences were synthesized, and annealed to create double-stranded inserts harboring the 20 bp gRNA target sequences, two BbsI restriction sites, 8 bp barcodes unique to each sgRNA while differed from each other by at least two bases, and 5' overhangs at their ends. To generate the pooled storage vector library, the 153 annealed inserts were mixed at equal ratios and cloned in the pAWp28 storage vector (digested with BbsI and Mfel) via a single pot of ligation reaction via their compatible ends. To build the barcoded sgRNA library, another one-pot ligation reaction was performed with the pooled storage vector library digested with BbsI, and an insert containing the gRNA scaffold sequence, BamHI and EcoRI restriction sites, and 5' overhangs at their ends that was prepared via synthesis and annealing of an oligo pair Sl and S2. The pooled storage vector and the barcoded sgRNA libraries were both prepared in Endura competent cells (Lucigen) and purified by the Plasmid Midi Kit (Qiagen). We confirmed that 97% (i.e., 32/33 E. coli colonies) of randomly picked colonies from the barcoded sgRNA storage pool harbored the correct gRNA target sequences via Sanger sequencing.
Pooled lentiviral vector libraries harboring single or combinatorial gRNA(s) were constructed with same strategy as for the generation of single and combinatorial sgRNA constructs described above, except that the assembly was performed with pooled inserts and vectors, instead of individual ones. Briefly, the pooled U6p-sgRNA inserts were generated by a single-pot digestion of the pooled storage vector library with BglII and MfeI. The destination lentiviral vector (pAWpl2) was digested with BamHI and EcoRI. The digested inserts and vectors were ligated via their compatible ends (i.e., BamHI + BglII & EcoRI + Mfel) to create the pooled wise sgRNA library (153 sgRNAs) in lentiviral vector. The one-wise sgRNA vector library was digested again with BamHI and EcoRI, and ligated with the same U6p-sgRNA insert pool to assemble the two-wise U6p-sgRNA library (153 x 153 U6p-sgRNAs = 23,409 total combinations). After the pooled assembly steps, the sgRNAs were localized to one end of the vector construct and their respective barcodes were concatenated at the other end. The lentiviral sgRNA library pools were prepared in XL10-Gold ultracompetent cells (Agilent Technologies) and purified by Plasmid
32