Effect of polymer chemistry on globular protein–polymer block copolymer self-assembly
14
0
0
Texte intégral
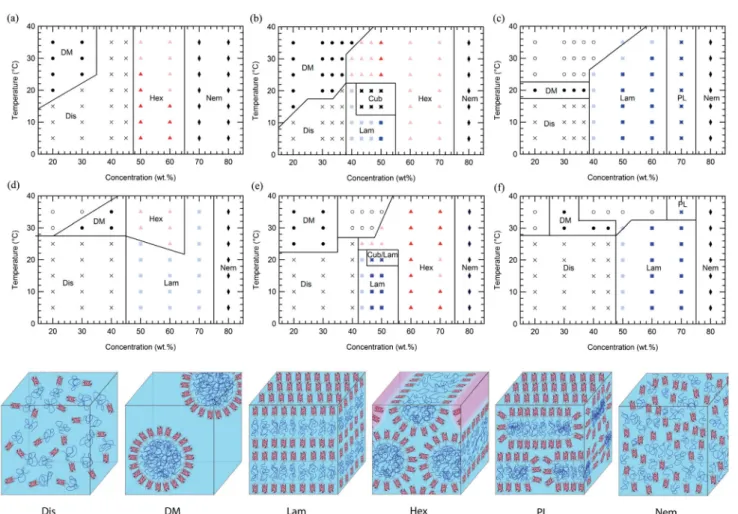
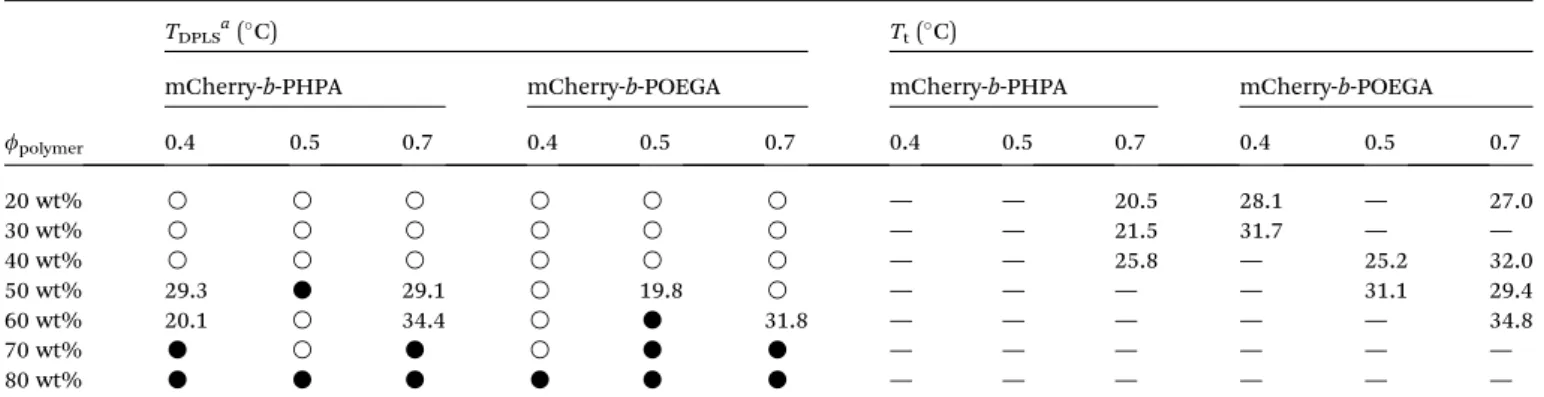
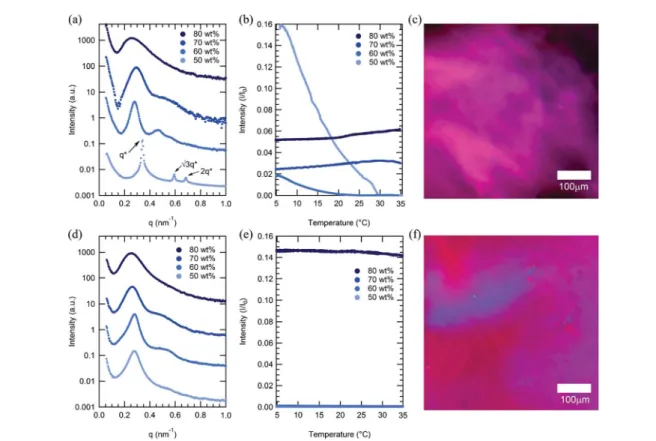
Figure
Documents relatifs
