Dissecting the molecular mechanisms of therapeutic
resistance in cancer
By
Vibhuti Agrawal
B.Tech in Biochemical Engineering and Biotechnology (2010)
M.Tech in Biochemical Engineering and Biotechnology (2011)
Indian Institute of Technology, Delhi, India
Submitted to the Department of Biological Engineering in Partial Fulfillment of the
Requirements for the Degree of
Doctor of Philosophy in Biological Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2017
02017 Massachusetts Institute of Technology. All rights reserved
Signature of the Author...
red.ac
...Vibhuti Agrawal Iepartment of Biological Engineering
Signature redacted
May9th 2017C ertified by ... ...
Forest M. White Professor of Biological Engineering
A ccepted by ...
ARCHIVES
MASSACHUSETTS INSTITUTE OF TECHNOLOGYJUL 122017
Thesis SupervisorSignature
redacted--...
Mark Bathe Associate Professor of Biological Engineering Chair, Graduate Program Committee of Biological EngineeringThis doctoral thesis dissertation has been examined by following thesis advisory committee members
Forest M. White, PhD
Thesis Supervisor
Professor, Biological Engineering Massachusetts Institute of Technology
Douglas A. Lauffenburger, PhD
Thesis Committee Chair
Professor, Biological Engineering, Chemical Engineering and Biology Massachusetts Institute of Technology
Robert A. Weinberg, PhD
Thesis Committee Member Professor, Biology
This thesis is dedicated to the loving memory of my mother:
my inspiration, my guiding light, and
Dissecting the molecular mechanisms of therapeutic resistance in
cancer
By Vibhuti Agrawal
Submitted to the Department of Biological Engineering on May 9th 2017 in partial fulfillment of
the requirements for the degree of Doctor of Philosophy
Abstract
Therapeutic resistance continues to be a persistent challenge in medical oncology. In clinical settings, resistance can occur at the beginning of treatment, or may be acquired after an initial clinical response to the therapy. Several mechanisms of drug resistance have been described in cancer, including alterations in the drug transport and metabolism process, mutations in drug-target, activation of bypass signaling pathways, inhibition of cell-death pathways, and induction of an epithelial to mesenchymal transition (EMT) in response to cytotoxic or targeted therapies. In this study, I have investigated the molecular mechanisms underlying ZEB 1-induced EMT and established a new computational framework that uses inter-animal heterogeneity to identify drivers responsible for variable phenotypic responses across different animals.
EMT describes a cell-state switching process wherein epithelial cells lose their tight cell-cell junction contacts, and acquire the ability to migrate and invade the surrounding stroma to enter
into blood circulation. Given the widespread role of EMT in drug resistance, it is imperative to identify therapeutic strategies to inhibit this transition. To identify druggable targets to block EMT progression, and therefore overcome EMT-mediated therapeutic resistance, I studied the effects of ZEB 1 expression on cellular signaling networks. By quantifying changes in tyrosine phosphorylation at different time points during ZEB 1-induced EMT, I found that Src family kinases (SFKs) were activated within 24 hours of ZEB 1 expression. Inhibition of SFKs blocked not only ZEB 1-induced EMT, but also EMT initiated by TGFp- and EGF signaling pathways in both breast and NSCLC cell-lines. SFK inhibition also prevented EGFR inhibitor-induced EMT and drug resistance in NSCLC cells both in vitro and in vivo. Mechanistically, SFK activation stabilized ZEBI by promoting ERK1/2-mediated phosphorylation on three serine residues, S583,
activity with regards to decreasing stability of ZEB 1 and inhibiting ZEB 1-induced EMT. These results provide a new therapeutic application of SFK inhibitors as a potential anti-EMT therapy, to enhance the susceptibility of cancer cells to chemo- or targeted therapies.
In the second part of this thesis, I have described a computational framework that leverages inter-animal heterogeneity to identify molecular mechanisms underlying variable phenotypic
responses across different animals. Substantial inter-animal variability in phenotypes within the same treatment group, limits our ability to draw conclusions or gain meaningful insights about a biological process by simply averaging the data. To identify molecular drivers for heterogeneous phenotypic responses, I have established a method where each animal is considered as an individual entity whose phenotypic response is dependent on the state of its underlying signaling networks. As a proof of concept, I have used this method to successfully predict the resistance mechanisms of CDK4/6 inhibitor, palbocilib in two GBM PDX and one MPNST PDX models. The GBM6 model activated EGFR signaling upon treatment with palbociclib whereas the GBM22 and MPNST3 models activated SFKs and PDGFRa signaling in resistant tumors. Across all three PDX tumor models, treatment with combination therapies, consisting of palbociclib and an inhibitor targeting the activated bypass signaling pathway, substantially prolonged survival of mice. Thus, these results suggest that inter-animal variability can be used as a tool to predict drivers for a specific phenotypic response across different treatment
conditions.
Thesis Advisor: Forest M. White, PhD
Acknowledgements
First and foremost, I would like to thank my thesis advisor, Prof. Forest White for his unwavering support and guidance throughout my graduate career. His warm laughter, enthusiasm and positive energy are contagious and instrumental in lifting up my spirits,
especially after a streak of failed experiments. I am extremely grateful to him for giving me the freedom to design my own project and pursue questions that interested me the most, while also providing his insightful thoughts and suggestions at every step during my PhD. As a great
mentor, he also helped me think through my career goals and forced me to ask myself difficult questions. I would also like to thank the members of my thesis committee - Prof. Doug
Lauffenburger and Prof. Bob Weinberg for their helpful advice and suggestions throughout my PhD.
This thesis wouldn't have been completed without the help and support of the members of White Lab. During the past five years, White Lab has become my second home due to its jovial,
cooperative, and positive environment. Moreover, White lab provides a perfect environment for interesting scientific discussions that have helped me become a better scientist. I am especially grateful to Dr. Amanda Del Rosario for teaching me how to run western-blots, answering my countless questions and above all, being a great scientific mentor who made me think beyond my experimental results. I would like to also thank Dr. Rebecca Lescarbeau for helping me get
started in the lab and teaching me all about the mass-spectrometry and tissue-culture. A big thanks to Dr. Antje Dittman for her everyday support, scientific and philosophical discussions
during my PhD as well helping me think about my future career goals. I would also like to thank Emily Whitehead for working with me to help me advance different scientific projects but more importantly giving me first-hand experience of being a trainer. A special thanks to Dr. Bryan Bryson for helping me set my professional goals, and to Dr. Kristina Emdal for proof-reading my thesis at the eleventh-hour. I am thankful to several other people -Jacqueline Ekins, Aaron Gajadhar, Elizabeth Gordon, Jose McFaline, Daniel Sears, Tim Curran, Daniel Rothenberg, Nader Morshed, Raven Reddy, Lauren Stofer, Yang Wang, Ishwar Kohale and Jacqueline Gerritsen - who have played a key role in making White Lab a fun-place to work.
I can't get through the first page of acknowledgements without thanking Dr. Brian Bierie from
guidance. Time and again, at every step of the project - be it designing the questions I want to ask, the model system I want to choose, troubleshooting my experiments, discussions on additional experiments that need to be done, or writing the manuscript - he helped me navigate my way through all of it. He has been a huge force in my graduate career; he has helped me
sharpen my scientific thinking in numerous ways, and pushed me to ask difficult questions. He is my go-to person for any questions about EMT, or molecular biology techniques in general.
I would also like to thank my collaborators outside MIT for providing timely guidance,
resources, and reagents. I would like to thank Dr. Nitin Shirole, Dr. Serif Senturk and Prof. Raffaella Sordella for providing me with the A549 cell-lines and helping me do in vivo experiments. I would also like to thank Dr. Fumi Kinose and Dr. Eric Haura for generously giving me their H1975 parental and the EGFR inhibitor resistant cell-lines. I am also grateful to Prof. Chris Sander and Dr. Nicholas Gauthier for their help with the MPNST samples, and teaching me the nuts and bolts of a good collaboration. Lastly, I am indebted to many people from Sarkaria Lab at Mayo Clinic, without whom I wouldn't have been able to investigate
inter-animal heterogeneity. My special thanks to Prof. Jann Sarkaria, Dr. Brett Carlson, Dr. Ann Tuma and Dr. Katrina Bakken for being great collaborators - they were instrumental in conducting all the mouse experiments involving GBM PDX tumors. I am indebted to Schlumberger Faculty for Future Foundation and Ludwig Center for Molecular Oncology for their generous fellowship awards. I am also grateful to NIH and NCI for supporting my research.
A good support systems is important for surviving the graduate school. I feel fortunate to be
surrounded by friends who stood by me through peaks and valleys. It is a pleasure for me to be a part of "Powerpuff Girls" group with Ankur, Pritika, Ranu and Ruchira. Ankur, being the other PhD in this group, always understood my predicaments and has been my personal, professional and technical counsellor. When it comes to designing experiments, she is one person who would always ask me about my negative and positive controls in any given experiment. I vividly remember one incidence out of many -I was stuck with my project and all my hypotheses were
failing. Not sure what to do next, I called up Ankur in the middle of the day. She patiently listened to the entire experimental designs, probed me for all the controls, and then, suggested me a few different experiments to rule out the remaining hypotheses. Her advice worked like a charm -within the next couple of days, I had figured out the problem and overcome the
roadblock in my project! I will never forget (hopefully!!) our phone conversations while making dinner or doing grocery, or our crazy road-trips. A big thanks to Ankur and Pritika for reading and reviewing my PhD/fellowship applications and thesis proposals for all these year. Pritika is an amazing person, and a great friend- we were room-mates in undergrad and since then, we have probably never moved apart (emotionally). From helping me practice for my interviews, or set career-goals, you have been always there as a supportive and caring friend. Thanks to all my friends at MIT -Nova, Souparno, Manish, Malvika, Swati, Noor, Vivek and many others, for making this graduate school a fun and enjoyable experience; words fall short while describing your value in my life. I would also like to thank all my school friends - Anukampa, Resham, Jyoti, Mamta, Sakshi and Kanul for always pushing me beyond my limits and helping me realize my true potential. Even though, we have grown up since our school days, and travelled across the world, it still feels like the old times whenever we meet and talk!
Last but not the least, I owe everything to my family - my mom, dad, taiji, tauji, buaji, phuphaji, Saumya, Shubham, Riddhi and Rishabh - without their firm support, I would not have been able to undertake this journey. Thank you so much for your unconditional love and support at every step of life. I have always known that irrespective of wherever I go, I can always go home to them without thinking twice. This thesis is dedicated to my mother, who is my role-model, my inspiration, and the voice in my head who helps me make distinction between right and wrong, who helps me stay focused and encourages me to traverse difficult paths --- I love you mom! A
special thanks to Sumit, for being my "bitter-half' for all these years, for patiently listening to my blabber, encouraging me through difficult times, and most-importantly, just always being there for me. 0
Table of Contents
A b stract ... 4
Acknowledgements ... 6
C hapter I
...
12
1.1 M echanisms of therapeutic resistance in cancer ... 15
1.2 Quantification of tyrosine phosphorylation signaling networks ... 30
1.3 Data-driven modeling techniques ... 31
1.4 M otivation and rationale for thesis design ... 35
1.5 R eferences ... 3 8
C hapter 2
...
46
2 .1 In tro d uctio n ... 4 8 2.2. M aterials and M ethods ... 50
2 .3 R esu lts ... 6 5 2 .4 D iscu ssio n ... 9 2 2 .5 R eferences ... 9 5 2.6 Supplementary Figures ... 99
C hapter 3
...
112
3 .1 In tro d uctio n ... 1 14 3 .2 M aterials ... 1 16 3 .3 R esults ... 12 2 3 .4 D iscu ssio n ... 14 3 3 .5 R eferences ... 14 7 3.6 Supplementary Figures ... 152C hapter 4
...
159
4.1 Summary and Conclusions ... 160
4.2 Future outlook ... 163
4.3 References ... 166
C hapter 5
... 167
5. 1. Study of mechanisms underlying SFK activation in response to ZEB I expression ... 168
5.2 Identification of E3-ligases that regulates ZEB I stability in a phosphorylation-dependent manner ... 172
List of Fiures
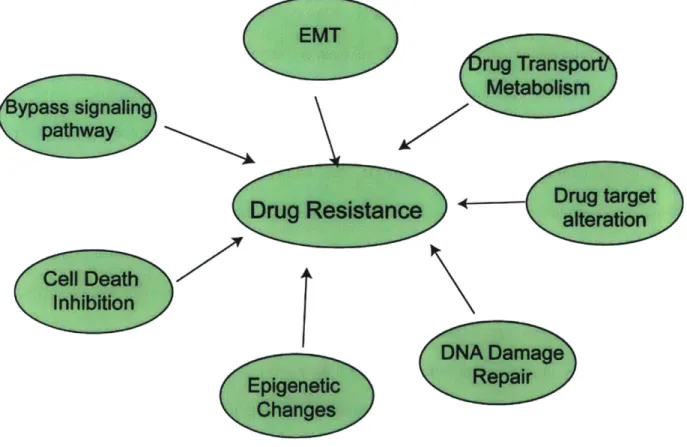
Figure 1.1 Mechanisms of drug resistance ... 15
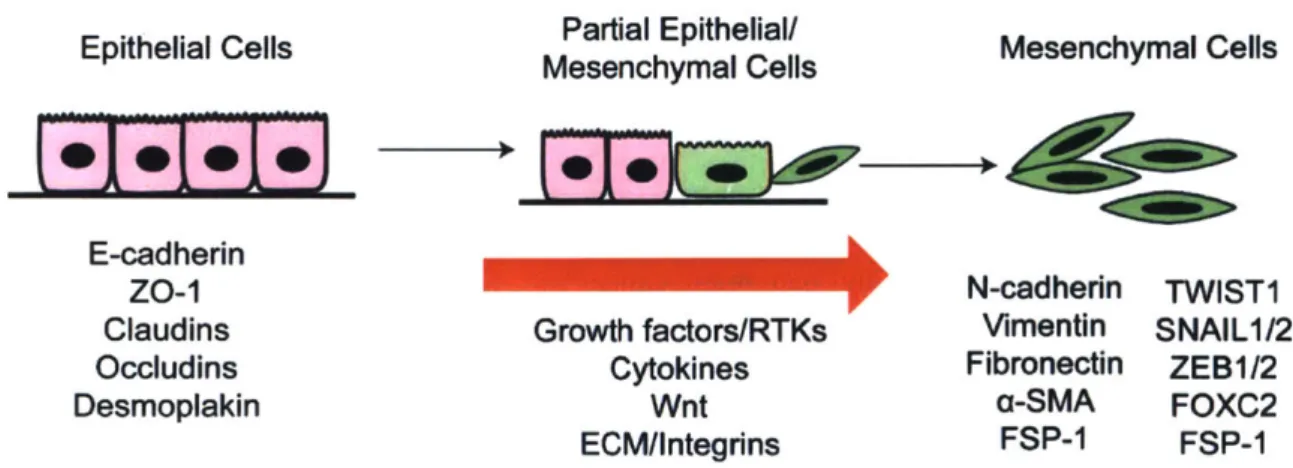
Figure 1.2 The epithelial to mesenchymal transition (EMT) in cancer progression ... 17
Figure 2.1: Characterization of temporal dynamics of ZEB 1 induced EMT in HMLEs...67
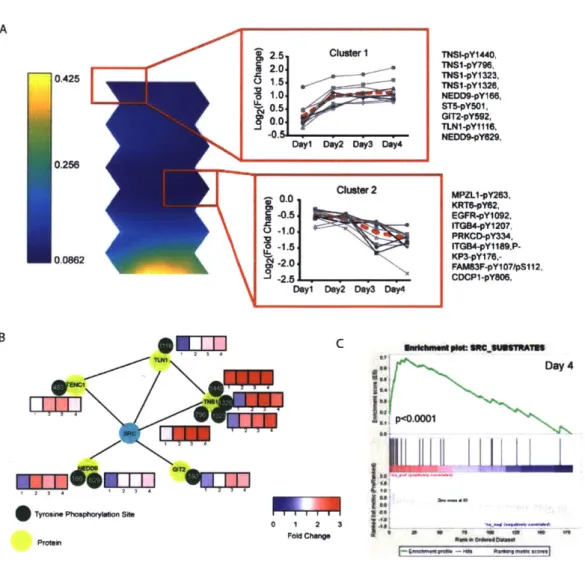
Figure 2.2: Src family kinases (SFK) are activated during EMT ... 69
Figure 2.3: Inhibition of SFKs is sufficient to block initiation of EMT ... 73
Figure 2.4: SFK inhibition prevents emergence of EGFR induced drug resistance in H 1975... 77
Figure 2.5: SFKs play a major role in regulating the protein stability of ZEB 1... 81
Figure 2.6: ZEB 1 stability is regulated by ERK1/2 activity ... 84
Figure 2.7: Phosphorylation on S583/S646/S679 augment ZEBI stability... 88
Figure 2.8: Src-family kinase and Erk MAPK activity stabilize ZEBI and are required for EMT pro gression ... 9 1 Figure 3.1: Identification of resistance mechanism to palbociclib in GBM6 PDX mouse model ... 12 5 Figure 3.2: Testing model prediction in vivo for GBM6 PDX mouse model ... 129
Figure 3.3: Identification of resistance mechanism to palbociclib in MPNST3 PDX mouse model ... 1 3 3 Figure 3.4: Identification of resistance mechanism to palbociclib in GBM22 PDX mouse model ... 1 3 7 Figure 3.5: Testing model prediction in vivo for GBM22 PDX mouse model ... 141
Figure 5.1 Role of ROS in ZEB 1-induced EMT ... 169
Figure 5.2 Identification of E3 ligase responsible for ubiquitinating and degrading non-phosphorylated Z E B 1 ... 173
List of Supplementary Figures
Figure S2. 1: Characterization of dox-inducible system in HMLE-ZEB 1, Related to Figure 2.1. 99 Figure S2.2: SFKs are activated within the first 24 hours of ZEB 1 expression and this SFK activation is not dependent on TGFp signaling pathway, Related to Figure 2.2... 100
Figure S2.3: Inhibition of SFKs blocks EMT induction across multiple systems, Related to F igure 2 .3 ... 10 2 Figure S2.4: Co-treatment with saracatinib decreases tumor growth by preventing TKI-induced EMT and enhanced killing of stem-cell subpopulation in H1975, Related to Figure 2.4 ... 104 Figure S2.5: Half-life of ZEBI is decreased due to SFK inhibition, Related to Figure 2.5 ... 106 Figure S2.6: Inhibition of MEK-ERK signaling pathway also blocks EMT induction across m ultiple system s, Related to Figure 2.6... 108
Figure S2.7: ZEBI phosphorylation is essential for EMT induction, Related to Figure 2.7... 110 Figure S3.1: Experimental design to characterize tumor heterogeneity in PDX mouse models, R elated to F igure 3 .1... 152
Figure S3.2: PLSR regression graphs for GBM6 PDX model, Related to Figure 3.1 and Figure
3 .2 ... 15 4
Figure S3.3: PLSR regression graphs for MPNST PDX model, Related to Figure 3.3 ... 156 Figure S3.4: PLSR regression graphs for GBM22 PDX model, Related to Figure 3.4 and Figure
Chapter 1
Cancer, the second-most leading cause of mortality worldwide, accounted for more than 8 million deaths and -14.1 million new cases in 2013. If the cancer incidence rates remain unchanged, the global cancer burden is expected to increase to 21.7 million new cases with 13 million deaths by 2030'.
Significant progress has been made in our ability to treat cancer over the course of last century. Until the 1950s, surgery and radiotherapy dominated the field of cancer therapy, however its success was limited by the emergence of metastases in distant organs2. At the end of second world-war, a number of drugs that inhibited DNA synthesis were approved for treating cancer, including folic acid analogues such as methotrexate, nitrogen mustard (mechlorethamine), and 5-fluorouracil3-6. Due to their efficacy in killing tumor cells, these compounds, also called
chemotherapeutic drugs, quickly gained popularity and became an integral part of cancer treatment. The next few decades witnessed the development of several additional drugs with similar mechanisms of action. However, the success of these drugs was short-lived as almost all patients relapsed and returned to clinics with a more aggressive tumor2. Meanwhile, with advances in our understanding of cancer biology and tumor progression, drugs specifically directed towards cancer cells were developed. These drugs, called targeted therapies, consisted of small molecule inhibitors or monoclonal antibodies against a specific protein that was important for driving tumor progression 7-1. While targeted therapies had fewer side-effects as
compared to the chemotherapy, the efficacy of these targeted therapies has also been limited by the emergence of drug-resistant tumor cells.
Development of resistance to chemo- or targeted therapies is a major challenge in the field of cancer treatment 2. Drug resistance in cancer can be categorized into two broad classes
resistance-mediating factors in the bulk tumor even before the therapy is administered. These resistant cells remain unaffected by therapy, thus making it ineffective. Adaptive drug resistance refers to the tumor cells that were initially sensitive to drugs, but have rewired their signaling networks in response to therapy to be able to survive the treatment. Recent studies have also shown that tumors are very heterogeneous in nature and thus, distinct sub-populations of cancer cells in bulk tumor can also exhibit resistance to therapy via completely separate molecular mechanisms13
Recent advances in genomic techniques have allowed us to rapidly identify alterations at a genomic scale across a large number of tumors, while advances in proteomic techniques have enabled identification of signaling pathways that are differentially regulated between sensitive
and resistant tumors14-19. This has led to identification of novel therapeutic targets and bypass signaling pathways that account for drug resistance in cancer cells. Moreover, use of high-throughput genomic techniques combined with computational data-driven approaches have also enabled identification of molecular signatures and genotypes that can predict response to drug treatments. Stratification of patients based on these molecular signatures have enabled better outcomes of clinical trials. Given the foreseeable development of resistance to these targeted therapies in cancer patients, study of resistance mechanisms has become an integral part of the
drug development pipeline for pharmaceutical companies. These studies are aimed at identifying novel drug targets that can be inhibited in combination with other chemo- or targeted therapies to overcome the problem of therapeutic resistance in cancer. The goal of my project was to study two resistance mechanisms in cancer - epithelial to mesenchymal transition (EMT) and
1.1 Mechanisms of therapeutic resistance in cancer
A diverse range of molecular mechanisms have been implicated in drug resistance in cancer.
These include alterations in drug transport (cellular influx/efflux rates) and metabolism, on-target
mutations that prevent a drug from binding and inhibiting its target, activation of bypass
signaling pathways providing compensatory inputs to the downstream components, phenotypic
switching by undergoing epithelial to mesenchymal transition (EMT), downregulation of
cell-death signaling pathways, and alterations in DNA damage repair pathways
12,20,21.
A role for
epigenetic changes and tumor microenvironment in therapeutic resistance is also being
increasingly recognized
21.Lastly, molecular and phenotypic heterogeneity inherent in all tumors
have also shown to contribute substantially to resistance
20EMT
rug Transpo
Metabolism
ypass signalin
pathway
~
rug ta
Drug Resistan
alterat
Cell Death
Inhibition
Epigenetic
Changes
Figure 1.1 Mechanisms of drug resistance
UNA
Damage
Repair
rget
a. Epithelial to mesenchymal transition
Epithelial to mesenchymal transition or EMT describes a series of biochemical changes during
which an epithelial cell changes its morphology, loses tight cell-cell junction contacts with
neighboring cells, and acquires the characteristics of a mesenchymal cell, such as the ability to
resist apoptosis and to invade the extra-cellular matrix to disseminate to distant organs in body.
At the molecular level, EMT is often described by decreased expression of epithelial markers
such as E-cadherin, claudins, and ZO- 1 and an increased expression of mesenchymal markers
including fibronectin, N-cadherin, and vimentin
22-27. EMT is a reversible process
-
mesenchymal
cells can further revert back to the original epithelial state by undergoing a mesenchymal to
epithelial transition (MET)
23,26,28EMT was first described by Dr. Elizabeth Hay in 1968 as a process indispensable for normal
embryonic development
29. Since then, EMT has been encountered in three distinct biological
settings that have different functional consequences, thereby providing a rationale for
classification of EMT into three sub-types
26.Type 1 EMT is associated with embryogenesis and
organ development as initially described by Dr. Hay. At different stages during gastrulation,
epithelial cells undergo EMT to give rise to mesoderm, endoderm, and mobile neural crest cells
which further give rise to the whole set of connective tissues including astrocytes, adipocytes,
chondrocytes, osteoblasts, and muscle cells. This type of EMT neither causes fibrosis, nor gives
rise to an invasive phenotype. Type 2 EMT is associated with wound healing, tissue regeneration
and organ fibrosis. In response to inflammation following physical injury or mechanical stress,
epithelial cells undergo EMT to generate fibroblasts and other cells to repair and reconstruct the
tissue after damage. Unlike type 1 EMT, type 2 EMT is associated with inflammation and it
such as during organ fibrosis, persistence of EMT results in an organ destruction. The last
sub-type, type 3 EMT has been observed in cancer progression and occurs in neoplastic cells that
have undergone genetic and epigenetic changes, especially in genes that favor clonal growth and
tumor development. In these cells, initiation of the EMT program gives rise to an aggressive
migratory phenotype, thus allowing them to intravasate into systemic circulation. Consequently,
these cells can undergo the reverse process of MET to seed micro- and macro- metastases at
distant organs
26. Importantly, EMT does not just describe the process of cells shifting between
two extreme states, epithelial and mesenchymal. Rather, EMT has been described as a
continuous and dynamic process, wherein cells can exist in a spectrum of intermediary phases,
also known as "metastable states". Cells in these partial EMT states express both epithelial and
mesenchymal genes to different extents, thus giving them the flexibility of becoming extreme
mesenchymal cells, or reversing back to epithelial cells depending on the environmental cues
23,
30
Epithelial Cells
Maea
l ells
Mesenchymal Cells
E-cadherin___________
zo-1
N-cadherin
TWIST1
Claudins
Growth factors/RTKs
Vimentin
SNAIL1/2
Occludins
Cytokines
Fibronectin
ZEB1/2
Desmoplakin
Wnt
a-SMA
FOXC2
ECM/Integrins
FSP-1
FSP-1
A common set of signaling pathways, transcription factors (TFs), epigenetic factors, and
microRNAs (miRNAs) are known to regulate all three types of EMT
25 26,
Intriguingly,
while the three described sub-types of EMT have distinct functional consequences, the specific
molecular signals differentiating them are not well-understood. For instance, inflammation
during trauma or injury is known to induce type 2 EMT to enable wound closure. Similarly,
inflammatory signals such as cytokines and immune cells present in tumor-microenvironment
play a major role in inducing type 3 EMT at the tumor edge
33. Notably, the signals that
distinguish EMT induction under both circumstances are unknown. Thus, more experiments are
needed to learn more about the similarities and dissimilarities among three sub-types of EMT.
In addition to cancer metastasis, type 3 EMT has been shown to play a major role in
development of resistance to chemo- or targeted therapies across different types of cancer
18,34-4Several studies show that cell lines, xenografts and patient samples that have decreased
E-cadherin expression, and gained vimentin and N-E-cadherin expression are resistant to EGFR
inhibitors
37-40,4. Furthermore, upregulation of classic EMT TFs has been observed in tumor
biopsies obtained from NSCLC patients who had developed resistance to EGFR inhibitors
18'
41.Recently two published studies have further established a role for EMT in mediating
drug-resistance to chemotherapy in breast and pancreatic cancer
35 36. Knock-down of EMT TFs, such
as TWIST 1 and SNAIL prevented growth of resistant tumors in a pancreatic cancer mouse
model
35. The mechanisms leading to EMT induction in tyrosine kinase inhibitor (TKI) treated
patients are not fully understood, and may involve activation of TGF-p signaling, or activation of
receptor tyrosine kinase (RTK) including IGF-lR, AXL, or Met
37,42-44,46.Increased
inflammation in the tumor-microenvironment has been associated with decreased tumor response
to EGFR inhibitors due increased TGF-p and IL6 signaling for survival
37. Activation of the RTK
AXL, through its overexpression or upregulation of its ligand GAS6, has also been proposed as a mechanism leading to EMT. In pre-clinical models, inhibition of AXL restored sensitivity to EGFR TKI4 2
-44. In the absence of secondary EGFR mutations, and amplification of AXL or
MET, the activation of IGF-1R can cross-talk with TGF-f-signaling pathway to induce EMT, thus driving resistance to erlotinib treatment in NSCLC46. Furthermore, differentiation of
mesenchymal stem-like cells to an epithelial cell state (i.e. induction of a MET program) has also been shown to sensitize cells to cancer therapy. Treating cells with forskolin or cholera toxin can induce a MET phenotypic switch in breast cancer cells by activation of protein kinase A (PKA) signaling47. Hence, a multitude of signaling pathways can regulate EMT-induced drug resistance in cancer cells, which makes it challenging to identify a single broad-spectrum anti-EMT
therapy.
In addition to different signaling pathways, EMT is also regulated by several TFs, epigenetic changes, miRNAs, alternative splicing events and extracellular signals including secreted soluble factors4 8. At the transcriptional level, a fine balance exists between TFs stabilizing the epithelial state and those stabilizing the mesenchymal state. Molecular reprogramming occurring during EMT is triggered by expression of various EMT TFs, including Snail family of zinc-finger transcription factors: SNAIl (Snail), and SNAI2 (Slug); the two-handed zinc-finger factors: 6EF1/zinc-finger E-box-binding homeobox (ZEB)1, and Smad-interacting protein (SIP)l/ZEB2; the basic helix-loop-helix factors: TWISTI, and TWIST2; E12/E47; Goosecoid; FOXC2; and
NFKB 23, 49-53. These TFs repress the expression of E-cadherin and induce the expression of
mesenchymal markers such as vimentin and fibronectin, thus allowing cells to dissociate from neighboring cells and increase their motility 54. Down-regulation of epithelial specific TFs such as grainyhead like protein 2 homologue (GRHL2), OVOLl/2, and erythroblast transformation
specific (ETS) TFs -ELF3 and ELF5, is also associated with EMT 5 . These TFs maintain
epithelial cell-state by providing a negative feedback loop to regulate the expression of ZEB 15 5, 5. To better understand the interaction between different TFs during EMT induction, Siletz et. al. built dynamic TF networks using a TF activity array 5. Although informative, this study also failed to identify good therapeutic targets to block EMT progression. In fact, one major drawback of studying TFs is that TFs are still considered undruggable. It is very difficult to block TF activity using a small-molecule inhibitor, which further limits their clinical relevance with respect to inhibiting them. However, these TFs can act as good biomarkers to identify different stages of EMT. Notably, this study highlighted the time-dependent mechanistic steps in EMT, which reinforces our notion that it is necessary to study temporal dynamics of EMT in greater detail to identify key players in this process.
While transcriptional regulation of EMT has been extensively studied, post-transcriptional, translational and post-translational regulators have also been highlighted in several studies. Integral to post-transcriptional regulation, miRNAs have emerged as potent regulators of EMT and MET through their ability to target the expression of key proteins regulating these processes. In general, miRNAs inhibit gene expression by cleaving target messenger RNA (mRNA) with subsequent degradation or translation inhibition 8' 59. Multiple miRNAs are known to target families of EMT TFs60-62. For instance, miR-200 family targets ZEB1/ZEB2 expression while
miR-34 family members tightly control the expression of SNAII63-66. Similarly, E-cadherin gene
expression is targeted by miR-9 while miR-194 inhibits EMT by repressing N-cadherin gene
676
expression 769. In addition to their role in decreasing translation of their direct mRNA targets, miRNAs can also cross-talk with epigenetic machinery to impact gene expression. For instance, increased SIRTI expression following activation of the TGF-P signaling pathway in mammary
epithelial cells can silence miR-200 promoter by histone deacetylation, resulting in increased
ZEBJ expression0. Also, loss of miR-200 leads to an increase in SUZ12 expression, which
results in polycomb-mediated repression of E-cadherin gene expression71. Various therapeutic strategies centered around miRNAs have been proposed. For instance, delivery of antisense oligonucleotide of miR-10b, a microRNA expressed under the control of TWIST, reduces the metastatic potential in a mouse mammary tumor model72. Similarly, overexpression of tumor suppressive miR-7 has been shown to inhibit EMT and rather promote MET by inhibiting
IGF-IR induced SNAIl expression73. In spite of these promising studies, there are still several challenges in using miRNAs as therapeutic tools in clinic to treat cancer patients. Some of these challenges include development of efficient systems to deliver miRNAs or antisense
oligonucleotides that target miRNAs, as well as improving the stability of these miRNAs or antisense oligonucleotides in physiological conditions65,7476
The epigenetic regulatory machinery, comprised of histone and DNA modifiers, also plays a major role in modulating EMT7 7
. SNAIl induces repressive histone modifications at the
E-cadherin promoter in the mesenchymal state by recruiting histone deacetylasel (HDAC 1) and
HDAC2. Treatment with the HDAC inhibitor, trichostatin A, blocks the repressive effect and
prevents metastasis7 8. Another mechanism by which SNAIL represses expression of target genes is by mediating the recruitment of histone methyltransferases SUV39H1 and G9a (also known as EHMT2), and DNA methyltransferases (DNMTs) 79. Similarly, ZEB 1 also cooperates with the deacetylase sirtuin 1 (SIRTI) at the E-cadherin promoter, leading to the deacetylation of histone
H3 and reduced binding of RNA polymerase II5048'80 . Furthermore, it has been shown that in the
epithelial state, the promoter region of ZEB 1 carries 2 methylation marks -H3K27me3 and H3K4me3. These two methylation marks on the ZEB 1 promoter not only repress its
gene-expression but also maintain the promoter in a poised state where it can readily respond to signals from the microenvironment such as TGFP. In response to cues, H3K27 methylation marks get removed, thereby resulting in active transcription of ZEB 181. This type of switching between the methylation marks may contribute to the reversible nature of EMT. A recent study has also highlighted a role for the histone demethylase, PHF2 in MET induction in cancer cells47. Given that epigenetic regulation of different genes plays an important role in cancer progression, a few inhibitors targeting HDACs, BET bromodomains, and histone demethylases have been developed and tested in clinic8 2. However, many of these drugs are broad-spectrum inhibitors that affect many proteins at once, thereby decreasing their clinical efficacy.
At the post-transcriptional level, there also exists an EMT-specific alternative splicing program which can modulate the cellular phenotype. Genes, like FGFR2, FGFR1, and STX2, which play a central role in inducing EMT are alternatively spliced in epithelial and mesenchymal states. Similarly, genes responsible for important cellular functions such as migration, adhesion and regulation of actin cytoskeleton have been shown to be expressed as differentially spliced isoforms during the EMT process8 3
At the post-translational level, alterations in signaling networks has been associated with EMT program. For instance, phosphorylation of specific tyrosine residues in proteins important for integrin signaling such as paxillin, p130Cas, Fak and Pyk2 drive cells towards EMT84. In another
study, by measuring changes in protein, phosphoprotein and mRNA transcript abundance, a shift in survival signaling and metabolic networks during EMT was demonstrated85. This study showed that while epithelial cells were dependent on EGFR and IGF- 1 R signaling for survival, mesenchymal cells were insensitive to their inhibition. Mesenchymal cells were instead
However, this study only investigated signaling networks in the initial epithelial and final mesenchymal states without providing any information about the mechanism underlying the change in survival signaling networks. Importantly, in order to identify anti-EMT therapies and inhibit the emergence of EMT-mediated drug resistance, a thorough understanding of molecular mechanisms underlying the transition process is indispensable. Studying dynamic changes in signaling networks during EMT will give us new insights into alterations occurring at different stages of EMT. This would enable us to identify therapeutic targets that can be inhibited to block emergence of EMT-mediated drug resistance in cancer cells.
Besides phosphorylation of several components of signaling cascades, phosphorylation of EMT TFs has been shown to regulate their sub-cellular localization, stability, and function. For instance, PAK1, or Src-ERK signaling axis, or LATS2-dependent phosphorylation of SNAIl promotes its nuclear localization and stability, therefore enhancing its ability to induce EMT689 On the contrary, GSK3p- or PKD1 -mediated phosphorylation of SNAIl decreases its half-life by promoting binding to E3 ubiquitin ligases such as
P-Trcp
and FBXO1 1 ligase, consequently reducing its ability to initiate EMT process90-91. ZEBI can also be phosphorylated and stabilizedby ATM kinase in response to DNA damage, which further mediates deubiqutination and
stabilization of CHK1, thereby promoting homologous DNA repair and resistance to radiation therapy9 2. Similarly, dysregulation of TWIST1 phosphorylation has been shown to hinder normal limb development and cardiac remodeling93' . Modulation of stability and function of EMT TFs by different kinases provides us with an alternative route to block the activity of these TFs by
using small-molecule inhibitors targeting specific kinases. Moreover, many of these inhibitors have already been approved by FDA and are used in clinic for treating cancer patients, which makes it easier to test their efficacy as potential anti-EMT therapies.
b. Activation of pro-survival signaling pathways
Cells sense changes in the extra-cellular environment via distinct RTKs present on their cell surface. These receptors signal through shared downstream effector proteins such as MAPK, PI3K/AKT, or mTOR to ultimately alter expression of different genes that can induce distinct cellular functions. Given that multiple RTKs signal through common downstream adapters and kinases, a common strategy by which tumor cells exhibit resistance to the inhibition of one RTK is through "bypass signaling", the parallel activation of another RTK that is not targeted by the drug. Thus, even in the absence of positive signals from the first driver RTK, the effector molecules continue to signal due to the inputs received from the second compensatory RTK.
In one of the earliest reported examples of bypass signaling, amplification of MET was shown to cause resistance to EGFR inhibitors in 20% of the NSCLC patients95. Upregulation of Met phosphorylation in these tumors resulted in activated PI3K/AKT signaling despite inhibition of EGFR 96. Subsequently, activation of IGF-1R, AXL, and PDGFR RTKs has been associated with resistance to EGFR inhibitors in clinic 9'. Signaling through other RTKs can also transactivate a receptor and amplify the signal downstream, thereby bypassing the effect of small-molecule inhibitors or antibodies targeting the specific receptor. Activation of EGFR and HER3 kinases, and increased heterodimers of EGFR/HER2 contributes towards resistance to trastuzumab, a monoclonal antibody blocking HER2 in breast cancer patients1 7. Activation of non-receptor
tyrosine kinases such as Src family kinases (SFK) have also been implicated in driving resistance to TKIs. For instance, YES 1, a member of SFKs was upregulated in lapatinib-resistant cells and co-treatment with a SFK inhibitor helped restore the sensitivity to lapatinib in breast cancer cells as well as xenograft tumors'7
loss of signaling resulting from targeted therapy, and the specific kinase utilized for this purpose is partially dependent on the tumor type and drug treatment.
Bypass signaling through altered RTK activity can be induced at multiple levels, including increased transcription, altered translation, altered degradation of the RTK, or increased autocrine signaling. One mechanism by which cancer cells increase autocrine signaling is by upregulating ADAM-mediated ligand shedding to enhance receptor activation 12. ADAM (a
disintegrin and metalloproteinase) enzymes are a family of membrane-associated metalloproteinases that can cleave and activate ligands for various growth factor RTKs.
Enhanced expression of ADAMs drives therapeutic resistance in cancer due to increased activity of a number of growth-factor RTKs9 8 . Moreover, inhibition of ADAM proteases has been
shown to impair EGFR signaling and sensitize cells to gefitinib treatment9 8. As ADAMs can regulate the shedding of ligands that activate numerous growth factor receptors, inhibiting its activity may have a more profound therapeutic effect than blocking individual growth factor receptors. However, ADAMs can also facilitate proteolytic shedding of several transmembrane receptors, which can further impact receptor activation, downstream signaling and drug response
101,102. A recent study has shown that cancer cells exhibited resistance to MEK inhibitors by
decreasing shedding of several RTKs including HER4, Met and AXL, thereby increasing their surface levels and mitogenic signaling 101. Thus, increased ligand and receptor shedding by ADAMs have an opposite effect on downstream signaling cascades in cells, thereby making it difficult to transition ADAM inhibitors to clinic effectively.
c. Tumor heterogeneity
Tumor heterogeneity has long been believed to be a critical determinant of therapeutic resistance
and patient outcome 20, 103-105. No two patients have tumors that are genetically, histologically and morphologically identical. Many host variables such as age, hormonal status, and genetic background, environmental factors, and importantly the tissue and cell of tumor origin, can introduce these inter-tumor differences 06. Tumor heterogeneity is not limited to differences between different patients, but can also be found within tumor harvested from a single patient1 05. Tumors are architecturally complex, differing regionally in vasculature, host infiltrates, and other characteristics that could have differential genotypic and phenotypic effects on the underlying cancer cells. Furthermore, tumors not only exhibit spatial heterogeneity but also temporal variation. Sampling different parts of the same tumors have revealed distinct genetic and epigenetic profile of the cancer cells at each site07' 108. Similarly, sampling of the tumor at different time-points during a disease progression has shown differences in the clonal composition of a given tumor 109, 110. Thus, tumors are extremely dynamic entities, evolving continuously in both space and time while following the classic Darwinian theory of "Survival of the Fittest"'1 1.
Advances in systems biology approaches including single-cell sequencing technologies have led to identification of distinct tumor sub-clones within a single patient pre-and post-treatment. Drug resistance can be ascribed to tumor heterogeneity in at least two ways - selection of a pre-existing resistant sub-clone in heterogeneous tumor or sub-clone specific upregulation of bypass signaling pathways to survive therapy. The most common mechanism of resistance to EGFR inhibitors is the acquisition of a secondary gatekeeper mutation in the ATP-binding pocket of the tyrosine kinase domain of EGFR (T790M)1 12, .13. Studies have shown the existence of a minor
sub-population of cells expressing the T790M mutation within untreated tumors"' "
Furthermore, presence of T790M mutation in the untreated tumors has a strong correlation with reduced progression-free survival in NSCLC patients"5. MET amplification is another known mechanism of resistance to EGFR inhibitors. High-throughput fluorescence in-situ hybridization
(FISH) analyses have been used to identify rare sub-clones of cells (<1%) with MET
amplification present prior to treatment initiation in lung cancer patients1 16.
Diversity in adaptive re-wiring of cellular signaling networks across individual tumor cells also plays a major role in emergence of drug resistance. A recent study used the combination of large-scale drug screening and whole-exome sequencing to show heterogeneity in resistance
mechanisms to erlotinib inhibition in a homogenous cell line"7. Clonally derived PC9 NSCLC cells were treated with erlotinib for -2 months and each individual resistant cell was expanded separately. Each of these erlotinib-resistant colonies demonstrated a different resistance
mechanism, many of which have been shown previously in clinic. Thus, even though the starting cell population for this study was clonally derived and can be assumed to be homogenous, the residual cells after erlotinib treatment had very different genetic and phenotypic features. Similarly, another study investigating mechanisms of erlotinib resistance in HCC827 NSCLC found that 60% of the sub-clones had MET amplification, while remaining 40% has undergone an EMT to exhibit resistance to erlotinib, suggesting induction of multiple resistance states from the same parental population 18. This heterogeneity in response to treatment presents a major
challenge in clinic; even if the combination therapies can be designed to eliminate one or few of the resistant colonies, there is no guarantee that these combinations will be effective in killing all resistant cells.
d. Others mechanisms of drug resistance
Tumor cells can develop resistance to chemo- and targeted therapy by several other mechanisms such as altering drug transport and metabolism properties, inducing on-target mutations, and dysregulating the DNA damage repair and pro-apoptotic pathway. Multi-drug resistance 1 (MDR1 also known as P-glycoprotein and ABCB 1) is a member of ABC transporter family that regulates flux across the cell-membrane119, 120. Recent reports have suggested that molecularly targeted therapies such as imatinib, erlotinib, sunitinib and nilotinib are also substrates for and modulators of MDR 20. MDR1 overexpression is associated with chemotherapy failure across
different cancers including kidney, colon, liver, prostate, lung, and breast cancer. Cancer stem-cells that are inherently resistant to therapies also have increased MDR1 expression12 1
. This would suggest that drug-efflux through MDR1 is a mechanism by which cancer cells display therapeutic resistance. However, MDR1 inhibitors, zosuquidar and tariquidar, have not been very successful in clinical trials in spite of their high potency and specificity. This failure can be partially explained by the possibility of functional redundancy across -49 members of ABC transporter family20' 122
Another mechanism of resistance to kinase inhibitors is the mutations in their gatekeeper residue in ATP binding pocket, such that they can no longer bind to the inhibitor. Prevalence of
EGFR-T790M accounts for 50% of the cases that have developed resistance to first generation EGFR
inhibitors, such as gefitinib and erlotinib15' . Second and third generation EGFR inhibitors have
been developed that can respectively target both WT-EGFR and EGFR-T790M, or mutant EGFR exclusively. Similarly, resistance to imatinib in chronic myeloid leukemia (CML) is due to mutation in gatekeeper residue of BCR-ABL1, T3151123. A new BCR-ABL1 inhibitor, ponatinib
BCR-ABLl inhibitors (such as dasatinib, nilotinib) failed to target. It will be interesting to see whether these third generation tyrosine kinase inhibitors also encounter target-associated mechanisms of resistance that are similar to the previous generation inhibitors.
Alterations in DNA damage repair (DDR) pathways can also result in tumors resistant to
chemotherapy. Cancers have frequently dysregulated their one or more DDR pathways either by mutations or epigenetic silencing of specific genes. This leads to complete dependence of cancer cells on the remaining intact repair pathways that otherwise are functionally redundant in normal cells, and thus can be targeted to induce synthetic lethality in cancer cells. However, resistance mechanisms develop by acquisition of new mutations that confer resistance to therapy, or restoration of initially dysregulated repair pathways by genetic reversion, or silencing of genes negatively regulating the repair pathway.
Increased expression of the drug target, or deregulation of proteins important for autophagy and apoptosis are other mechanisms that have been linked to resistance to chemotherapy and targeted therapies12.
Given the high incidence rate of therapeutic resistance in cancer patients, there has been an on-going extensive effort to characterize resistance mechanisms for different cancer treatments across distinct types and sub-types of cancer. In addition to profiling of genomic alterations in tumor samples to identify genetic alterations characteristic of therapeutic resistance, systems biology approaches have enabled identification of alterative signaling pathways that can also contribute to drug resistance. In this study, I have used large-scale and unbiased tyrosine phosphoproteomic profiling to delineate the molecular mechanisms underlying ZEB 1-induced EMT, as well as define novel combination therapies for resistance to CDK4/6 inhibitor in
1.2 Quantification of tyrosine phosphorylation signaling networks
Since the discovery of tyrosine kinases nearly 30 years ago, tyrosine phosphorylation (pTyr) has emerged as a fundamentally important mechanism for regulating many biological processes including cell proliferation, migration and survival25' 126. Perturbations in pTyr signaling
networks underlie many human diseases, including cancer127
-129. This has led to the development
of many tyrosine kinase inhibitors, several of which have been approved for clinical use 0. Several methods have been established to detect and quantify pTyr on a single protein or multiple proteins at once. Basic immunoblotting with phosphosite-specific antibodies for the target protein has been the standard approach for many decades. Recent development of high-throughput techniques, such as protein arrays, reverse-phase protein array (RPPA), and bead-based luminex assay (Luminex xMAP) have further enabled simultaneously monitoring of hundreds to thousands of proteins in a quantitative mannerm-m31 . These techniques are all based on the recognition of a phosphorylation site on the target protein by a phosphosite-specific antibody. There are a couple of major challenges associated with these techniques. First, the availability of a good phosphosite-specific antibody for a specific application, against the protein of interest, is the foremost limiting factor for these affinity-based assays. The second major limitation of these assays is the obligation to know before-hand the identity of proteins or signaling pathways that would be playing an important role in the biological process under investigation. This further restricts our ability to use these methods for discovery-based proteomics research in order to associate new proteins, or signaling cascades with a given phenotype.
Technical developments in mass spectrometry (MS)-based phosphoproteomics have enabled simultaneous quantification of hundreds of pTyr peptides without the need for a phospho-site
specific antibody for any given target or a priori knowledge of target proteins. Coupled with motif enrichment strategies, and metabolic and chemical labelling techniques, MS-based phosphoproteomics have become a standard technique for studying dynamic alterations in pTyr signaling networks. MS-based pTyr profiling has been successful in unraveling many different aspects of biology including mapping of downstream signaling pathways for various RTKs, identification of kinase-substrate motifs and delineation of novel mechanisms of therapeutic
134
resistance in several diseases including cancer'.
Despite many advantages of MS-based phosphoproteomics, there are a few inherent limitations of this technique. Due to the complexity and low abundance of pTyr peptides, extensive sample-preparation, enrichment techniques and high-resolution mass spectrometers are required to achieve substantial coverage of the phosphoproteome, thereby making these approaches time-and cost-intensive. In addition, interpretation of the "high-content" phosphoproteomics data is not simple and straightforward; extensive computational modeling is required to draw
biologically meaningful conclusions from these large-scale and unbiased phosphoproteomic datasets. In the section below, different data-driven modeling strategies have been discussed that are being widely used to gain insights from large "-omics" datasets.
1.3 Data-driven modeling techniques
Advances in "-omics" technologies have created a need for computational approaches to comprehensively query the massive datasets for interesting trends and patterns. Several clustering and enrichment tools developed originally for use with genomics datasets have also been applied to better understand the proteomics datasets. These computational techniques include both unsupervised clustering algorithms such as hierarchical clustering, k-means
135
such as partial-least squares regression (PLSR) and multi-linear regression 3. In addition to clustering quantitative phosphorylation profiles, several approaches that build a network map have also been used to analyze phosphoproteomics datasets. Many of these network building approaches rely on protein-protein interaction databases that often do not contain information about the post-translational modification of a given protein, thus limiting our ability to represent site-specific information on these network maps or to draw conclusions based on
phosphorylation of a given protein. Database-agnostic network map building techniques such as partial correlation network analysis have been developed to alleviate this challenge. However, they do not provide much information about the proteins or phosphorylation sites that have not been measured experimentally. Thus there are several approaches to analyze phosphoproteomics data, and the choice of method is dependent on the questions being asked as well as the different experimental conditions. Next section presents a detailed description of the four data-driven modeling techniques that have been used in this study to analyze pTyr data.
a. Self-Organizing Maps
A self-organizing map, also known as SOM algorithm or Kohonen Map, is an unsupervised
neural network based on a competitive learning clustering algorithm 136. It projects high-dimensional input data onto a low high-dimensional (usually two-high-dimensional) space. The algorithm works by first choosing an output neuron that most closely matches the presented input pattern, then determining a neighborhood of excited neurons around the winner, and finally, updating all of the excited neurons. This process iterates and fine tunes itself until all the data-points have been included in the map, and thus it is called self-organizing. The outcome weight vectors of the
Analysis of pTyr data with self-organizing maps has previously shown to identify clusters of peptides with distinct temporal responses to different stimulation conditions 128, 137. A closer
examination of different clusters further led to generation of new hypotheses with better insights into molecular mechanisms. In this study, I have used SOMs to identify clusters of peptides that have similar phosphorylation dynamics during ZEB 1-induced EMT. A detailed inspection of these clusters highlighted key nodes that can be targeted to inhibit ZEB 1-induced EMT.
b. Principal Component Analysis
Principal Component Analysis (PCA) is another unsupervised clustering algorithm that uses principal components to identify the linear combinations of signaling axes most tightly connected with one another. Furthermore, principal components function as super-axes, and allow us to view the entire data space in fewer dimensions that capture the most important information in each of the original signaling axes. These principal components are defined by providing weight to signals with high covariance and de-emphasizing signals that show little covariance with other signals. In this way, PCA condenses measurements to highlight the global patterns in the data set as reflected by the appropriate number of dimensions, typically 2 or 3 that capture the maximal covariation between all of the signals.
PCA has proven to be a valuable tool for visualization of high-dimension data spaces for a
number of biological applications 135, 138 PCA has been shown to qualitatively discriminate
apoptotic cell fates based on measured signaling profiles13 9. PCA was also used to classify hematological cancer cells according to their pathological origin based on their signaling network states140. In systems biology, PCA has also been coupled with sensitivity analysis to help reduce the complexity of mechanistic signaling models41. In this study, I have used PCA to
c. Partial Least Squares Regression
While SOM and PCA were unsupervised clustering algorithms, partial least squares regression
(PLSR) is a supervised learning algorithm. PLSR models the relationship between
experimentally determined cell signaling and phenotypic responses. While PCA is useful in identifying signaling variables that define the maximum variance in the data, PLSR is valuable for generating hypotheses wherein some variables are causally related to other variables. In other words, PLSR defines variables as dependent and independent and then identifies a linear solution that could relate the dependent variables to independent variables. In the past, PLSR models built using cellular signaling networks and cell migration (phenotypic response) were successful in identifying signals governing cell migration 42. Similarly, PLSR modeling can be used to identify
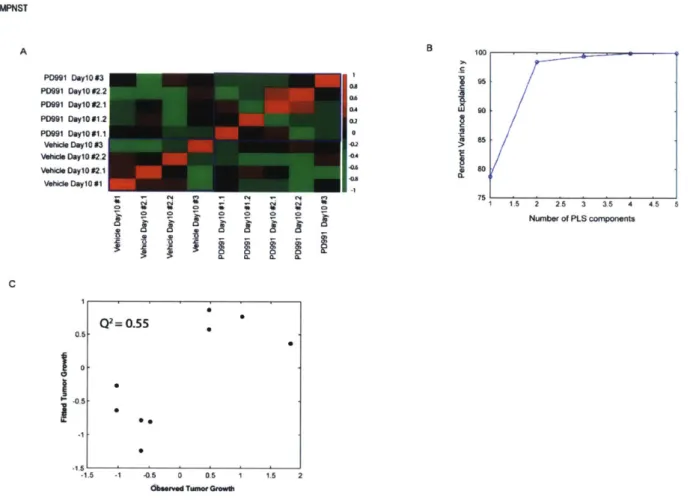
most important determinants of cell proliferation or tumor growth43. In this study, I have applied the PLSR algorithm to predict novel resistance mechanisms for a cell-cycle inhibitor in GBM and MPNST.
d. Partial Correlation Network Analysis
Partial correlation network is an undirected graph wherein the edges represent partial correlation between two nodes after conditioning on all the other nodes. Thus, the strength of an edge is indicative of the ability of two nodes to predict each other as observed in multiple regression. This analysis can be used to build a network map without any prior knowledge about the protein-protein interactions from any database. Partial correlation networks have been used to identify regulatory changes in one or more pathways using single cell expression datasets144, or
microarray datasets for different diseased states45. In this study, I have used partial correlation network analysis in conjunction with PLSR to identify resistance mechanisms for CDK4/6 inhibitors in GBM and MPNST.
1.4 Motivation and rationale for thesis design
The main objective of this thesis was to investigate molecular mechanisms underlying therapeutic resistance in different types of cancer. Given that EMT and activation of bypass signaling networks has been described as two prominent resistance mechanisms across different types of cancer, I decided to investigate them further in great detail during my PhD thesis project.
Extensive studies over the last decade have shown that cancer cells exhibit resistance to chemo-or targeted therapies by undergoing EMT. TWIST, ZEB 1 and SNAIL family of TFs have been identified as the master regulators of EMT26, 146. However, like most other TFs, these proteins at
present remain non-druggable or are very difficult to inhibit, thus hindering our ability to block EMT. Importantly, a thorough understanding of the regulatory mechanisms modulating EMT would enable the identification of secondary druggable targets that can be activated or inhibited to inhibit EMT. A significant amount of work has been done to examine changes in expression profiles of different mRNAs or miRNAs, or alterations in human epigenome during EMT2 3,62,7 7,
147. However, these studies haven't resulted in identification of druggable targets to prevent EMT
induction. Few studies have used large-scale proteomics to investigate the role of different serine/threonine kinases and signaling networks during EMT, and yet most of the latest
generation of molecularly targeted cancer therapeutics are kinase inhibitors. Notably, these few protein-centric studies have primarily looked at only the start and end states of the EMT process
(i.e. epithelial and mesenchymal states), thereby providing little knowledge about the dynamic molecular mechanisms that drive EMT.
To address these unanswered questions in the field, I have studied temporal changes in the tyrosine phosphoproteome during ZEB 1-induced EMT in the first part of this thesis. I found that SFKs are activated within 24 hours of ZEBI overexpression. Inhibition of SFKs using small-molecule inhibitors is sufficient to inhibit ZEB 1-induced EMT, and block ZEB 1-mediated drug resistance in vitro and in vivo. Importantly, I established that SFK inhibition destabilizes ZEB 1
by preventing ERK1/2-mediated ZEBI phosphorylation on three serine residues - S583, S646
and S679. Mutation of all these three residues to a non-phosphorylated amino-acid, alanine, decreased ZEB 1's stability and prevented initiation of EMT, suggesting that ZEB 1
phosphorylation by SFK-ERK signaling axis is important for its ability to induce EMT.
The second part of this thesis deals with development of a framework that leverages inter-animal tumor heterogeneity to define novel combination therapies. A standard in vivo experimental design for identification of novel resistance mechanisms to a given drug involves three parts
-first is the injection of tumor cells or xenografts in 5-10 animals, second is to treat the animal for a defined number of days with the given drug, and the last step involves genetic and/or
proteomic profiling of these drug resistant tumors. Frequently, data from multiple animals within a single treatment group is combined and averaged, and then the corresponding averages are compared across treatment groups. However, substantial inter-animal heterogeneity within each treatment group often makes these comparisons across multiple treatment groups statistically insignificant, thus limiting our ability to make robust predictions about the resistance
mechanisms. These inter-animal variations are observed in both phenotypic data such as growth-rate or tumor volume as well as their core signaling network states. These variations from one
animal to another could result from many different causes, including differences in vascularization, innate immune response, tumor micro-environment, and/or intra-tumor heterogeneity.
In chapter 3, using inter-animal heterogeneity, I have described a new computational framework to identify bypass signaling resistance mechanisms across different cancer types. In this
approach, growth rates for each individual tumor is assumed to be dependent on the state of its own underlying signaling network. As a proof of concept, I have tested this approach in vivo across three distinct patient-derived xenograft (PDX) models that were treated with a cell cycle inhibitor, palbociclib. This mathematical framework successfully identified resistance
mechanisms for palbociclib treatment across two GBM PDX models and one MPNST PDX model. Remarkably, treating animals with the combination therapies as predicted by the model resulted in a significant improvement in survival of the mice and a reduction in tumor growth-rates. This approach of modeling phenotypic responses at an individual animal can be further extended to identify key driving proteins across different pathophysiological conditions.