Development of New Transition Metal Catalysts for C-N Bond Formation
and Continuous Flow Processes for C-F Bond Formation
By
Nathaniel H. Park
B.S. Chemistry B.S. Environmental Science Montana State University (2009)
ARCHIVES
MASSUETS ILNSTITUTEOF TECHNOLOGYNOV
09
2015
LIBRARIES
Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY IN ORGANIC CHEMISTRY
at the
Massachusetts Institute of Technology September 2015
0 2015 Massachusetts Institute of Technology All Rights Reserved
Signature of Author:
Sig
nature redacted
Certified by: Accepted by: Department of Chemistry July 07, 2015
Signature redacted
Stephen L. Buchwald Camille Dreyfus Professor of Chemistry Thesis SupervisorSignature redacted
Robert W. Field Haslam and Dewey Professor of Chemistry
This doctoral thesis has been examined by a committee of the Department of Chemistry as follows:
Professor Timothy M. Swager:
Professor Stephen L. Buchwald:
Professor Mohammed Movassaghi:
"I
Signature redacted
Thesis Cork ittee Chair
Signature redacted
Thesis Supervisor
Signature redacted
Z-1Development of New Transition Metal Catalysts for C-N Bond Formation
and Continuous Flow Processes for C-F Bond Formation
By
Nathaniel H. Park
Submitted to the Department of Chemistry on July 07, 2015
in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry at the Massachusetts Institute of Technology
Abstract
The work presented in this dissertation addresses the development of new methodologies and processes to form carbon-nitrogen (C-N) and carbon-fluorine (C-F) bonds. The development of methods for the formation of C-N and C-F bonds are highly important to chemistry in general and find broad application in many different areas of research. With regard to C-N bond formation, the development of new nickel and palladium catalyst for C-N cross-coupling is presented. Finally, the development of a new process to enable the rapid preparation of aryl fluorides via the Balz-Schiemann reaction is explored.
Chapter 1. Development of an Air-Stable Nickel Precatalyst for the Amination of
Aryl Chlorides, Sulfamates, Mesylates, and Triflates.
A new air-stable nickel precatalyst for C-N cross-coupling is reported. The developed catalyst system displays a greatly improved substrate scope for C-N bond
formation to include both a wide range of aryl and heteroaryl electrophiles and aryl, heteroaryl, and alkyl amines. The catalyst system is also compatible with weak base, allowing for the amination of substrates containing base-sensitive functional groups.
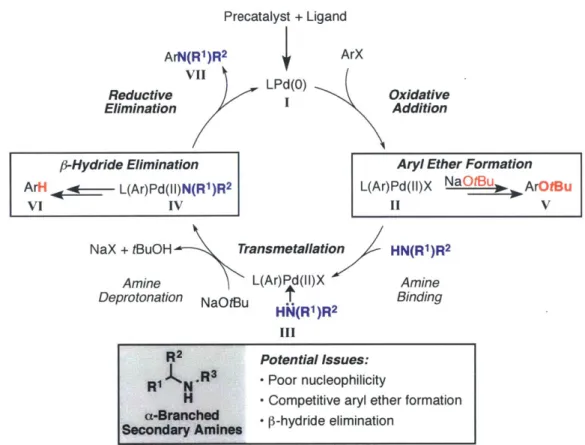
Chapter 2. Design of New Ligands for the Palladium-Catalyzed Arylation of a-Branched Secondary Amines.
In Pd-catalyzed C-N cross-coupling reactions, a-branched secondary amines are difficult coupling partners and often produce low yields of the desired product. To provide a robust method for accessing N-aryl a-branched tertiary amines, new catalysts have been designed to suppress undesired side reactions often encountered when
array of sterically encumbered amines, highlighting the importance of rational ligand design in facilitating challenging Pd-catalyzed cross-coupling reactions.
Chapter 3. Rapid Synthesis of Aryl Fluorides in Continuous Flow via the
Balz-Schiemann Reaction.
The synthesis of aryl fluorides (ArF) is of critical importance for the development of new and potent pharmaceuticals and agrochemicals. While there have been numerous and intense research efforts focused on developing new fluorination methods, the Balz-Schiemann reaction remains a valuable and efficient means of aryl
C-F bond construction from a vast pool of available aryl amines. However, the harsh
reaction conditions, modest yields, and often prohibitive safety concerns have limited the general application of this methodology. Here, we have developed a semi-flow process that enables safe handling of the potentially explosive aryl diazonium salt intermediates as well as improved yields of the desired aryl fluoride products. This process has been demonstrated on an array of different aryl and heteroaryl amine substrates containing a variety of different functional groups.
Thesis Supervisor: Stephen L. Buchwald Title: Camille Dreyfus Professor of Chemistry
Acknowledgements
Graduate school has been a singularly momentous and enormous undertaking. Having finally arrived at the end, I can safely say that did not make it this far based solely on my own sheer will and determination. After six years, three different cities, two different PhD advisors, and almost innumerable colleagues, there is no shortage of people whose help and support I am extremely grateful for.
First and foremost I'd like to thank both of my advisors, Profs. David Gin and
Stephen Buchwald. It was through their guidance and support that I have grown professionally and become a much better chemist. Dave was a great mentor whose enthusiasm for chemistry was very contagious and uplifting. His untimely passing was perhaps one of the most difficult times during my PhD studies. Thankfully, Steve not only made my transfer to MIT incredibly easy, but also accepted me into his group as a graduate student. During my time in his lab he has been very supportive and has given me the opportunity to work on many great projects. I have learned a lot from both Steve and Dave and I am very thankful to have worked for both of them.
I would also like to thank my thesis chair Prof. Tim Swager. Over the years Tim
has been a great source of insight for research and career development and I am very grateful for his support over the years.
I would be remiss if neglected to acknowledge my fellow colleagues from Gin lab
and the Tri-Institutional Program in Chemical Biology in New York City. They were a great group of people to work and hang out with. Particularly, I'd like to thank my bay mate Rashad Karimov for being an awesome person to work with and for helping me
get situated in the Gin lab. I'd also like to acknowledge my other Gin lab coworkers: Bryan Cowen, Yuan Shi, Alberto Fernandez-Tejada, Sudeep Prajapati, Eric Chea,
Jeremy Wilmot, Lars Nordstrom and others. They were a fantastic group of people to work along side during my time in the Gin Lab.
After transferring to MIT, it took quite a bit of time for me to actually enjoy doing chemistry again. Thankfully, there were many people who were friendly, welcoming, and helped me make the transition to the Buchwald lab. I was fortunate to be part of a great class of fellow Buchwald grad students in the group of eight: James Colombe, Katya Vinogradova, Mingjuan Su, Nootaree Niljianskul, Phil Milner, Nick Bruno, and Rong Zhu. They have been all been awesome to work and hang out with over the last few years. I've also had numerous fun times with my other fellow grad students, specifically:
Spencer Shinabery, Georgiy Teverovskiy (Team Nickel), Pedro Arrechea (Team Computer), Paula Ruiz-Castillo, Yang Yang, and Yuxuan Ye. During the last part of my studies I overlapped with many of the newly minted first-year grad students, namely: Jeffrey Yang, Anthony Rojas, Bryan Ingoglia, and Saki Ichikawa. Best of luck to all of you on the path ahead!
During my time in Buchwald lab I've been very fortunate to work alongside many, many talented postdocs that have come through the lab over the years. The problem with postdocs is that their time here is typically short and before you know it they have
moved on. Some of the first postdocs I overlapped with and learned a lot from were Andrew Parsons, Naoyuki Hoshiya, Mao Chen, Natalia Chernyak, Robb DeBergh, Jean-Baptiste Langlois, Matthias Oberli and Hong Geun Lee. Nate Jui and Alex Spokoyny
were two great guys who would always be willing to help answer a question or give me
feedback on a presentation. Sean Smith, Tom Barton, Yiming Wang, and Ye Zhu were a pleasure to work with and always fun to talk to. The latest crop of postdocs including Thierry Leon Serrano, Michael Pirnot, Jeff Bandar, Vasu Bhonde, Esben Olsen, Kurt Armbrust, and Sandra King have all made the lab a great place to come to work everyday. Kashif Khan, John Nguyen, and Stefan Roesner have been great friends as well as helped me immeasurably with my job interview preparations, including to listening to some of my presentations. Tim Senter also deserves a big debt of gratitude for his continuing help with the Balz-Schiemann project, being a great source of knowledge on all things relating to medicinal chemistry, and for listening to my job talk multiple times. I would also be remiss if I failed to mention that Christine Nguyen has
been practically running the lab for years and probably one of the kindest people I have met during my time here. I also owe a big thanks to Aaron Sather for fearlessly reading all of my papers, including the unpublished parts of this thesis, as well as giving
feedback on several of my talks. He has also been an all-around awesome colleague
and friend.
I would also like to give a big acknowledgement to my undergraduate advisor,
Hien Nguyen. Hien has been a great friend and mentor since I originally joined his lab
many years ago. The time I spent working for Hien is what prompted me to pursue a PhD in the first place and apply to schools that I would have otherwise considered
throughout my PhD, especially during the rough times following the passing of Dave and
I am extremely grateful for everything that he has done.
Finally, I would like to thank my parents and sister for their constant support throughout all of the ups and downs of graduate school. Graduate school would have been infinitely more difficult without their support and for reminding me that its important to take breaks from lab. Overall, I'd be hard pressed to say that my graduate school experience was great or easy; it tended to often be neither of those. However, I did learn a lot, become a much better chemist, and most importantly, met and befriended some awesome people along the way. Thanks again to you all!
Preface
This thesis has been adapted from the following published articles co-written by the author:
Park, N. H.; Teverovskiy, G. T.; Buchwald, S. L. "Development of an Air-Stable Nickel Precatalyst for the Amination of Aryl Chlorides, Sulfamates, Mesylates, and Triflates"
Org. Lett. 2014, 16, 220-223.
Park, N. H.; Vinogradova, E. V.; Surry, D. S.; Buchwald, S. L. "Design of New Ligands for the Palladium-Catalyzed Arylation of a-Branched Secondary Amines" Angew. Chem.
Respective Contributions
This thesis contains work that is the result of collaborative efforts between the author and other colleagues at MIT. The specific contributions of the author are detailed below.
The work in Chapter 1 was a collaborative effort between Dr. Georgiy Teverovskiy and the author. Dr. Teverovskiy performed the initial experiments, catalyst development, and initial substrate scope for this project. The author conducted all of the work presented in Chapter 1.
The work in Chapter 2 was a collaborative effort between Dr. David Surry, Dr. Ekaterina V. Vinogradova, and the author. Drs. Surry and Vinogradova performed initial experiments for this project. The author conducted all of the experiments in this chapter.
The work in Chapter 3 is currently a collaborative project between Dr. Tim Senter and the author. The author initiated the project, developed all of the reaction conditions, and is responsible for all the synthetic and spectroscopic data presented in this chapter.
Table of Contents
Intro d u ctio n ... . .. . ... 12 Chapter 1. Development of an Air-Stable Nickel Precatalyst for the Amination of Aryl Chlorides, Sulfamates, Mesylates, and Triflates.
1.1 Introduction... 19
1.2 Results and Discussion... 20
1.3 Conclusions... 27
1.4 E xpe rim e nta l... 2 7
1.5 R efe re nces... 5 9 1 .6 S p e ctra ... 6 3 Chapter 2. Secondary 2.1 2.2 2.3 2.4 2.5 2.6
Design of New Ligands for the Palladium-Catalyzed Arylation of a-Branched Amines.
Intro d u ctio n ... 1 17 Results and Discussion... 119 C o nclus io ns ... 12 6 E x pe rim e nta l... 12 6
R efe re nce s ... 18 7 S p e ctra ... 19 3
Chapter 3. Rapid Synthesis of Aryl Fluorides in Continuous Flow via the
Schiemann Reaction.
3 .1 Intro d u ctio n ...
3.2 Results and Discussion... 3 .3 C o nclus io ns ... 3 .4 E x pe rim e nta l... 3 .5 R e fe re n ce s ... 3 .6 S p e ctra ... Balz-315 326 331 332 357 362 C u rricu lu m V itae ... 390
Introduction
The synthesis of aryl carbon-nitrogen (C-N) and carbon-fluorine (C-F) bonds
are of critical importance due to their prevalence in natural products, pharmaceuticals,
and organic materials (Figure 1).) As such, the advancement of efficient methods or
processes for the construction of aryl C-N and C-F bonds have found broad application
in both academic and industrial research settings.2
Pharmaceuticals Natural Products Organic Materials
CF3 Me
- NHO
F 2 N Mel- Me
Me
Sitagliptin ( )-Murrayazoline CPD
Anti Hyperglycemic Hole-Transport Material
Merck
Figure 1. Selected compounds containing aryl C-N and C-F bonds.'
Transition metal catalysis C-N cross-coupling is a highly effective method for the
construction of aryl C-N bonds and has undergone significant development since its
initial report.3 A variety of transition metals have been used, however, most systems rely
upon Cu, Ni, or Pd as the metal. Catalyst systems that utilize Ni or Pd typically follow the general M(O)/M(II) catalytic cycle as depicted in Figure 2.3 However, Ni may also participate in a M(I)/M(Ill) pathway due to its ability to readily access multiple oxidation states.4 In the proposed M(O)/M(II) catalytic cycle, the generation of the catalytically active LnM(O) (I, Figure 2a) species is the entry point of the catalytic cycle. While there
are many readily available sources of Pd and Ni (e.g. Pd(OAc)2, Pd2dba3, Ni(COD)2,
NiCI2(PPh3)2), they often suffer from the need to be reduced (Pd(OAc)2, NiCI2(PPh3)2)
5
or pre-ligated (Pd2dba3)6 to generate the catalytically active species. These processes
reducing agent to be added such as phenyl boronic acid.7 In contrast, the development of precatalysts for Ni and Pd have greatly improved both the rapid generation of the active catalyst and the operational simplicity of the reaction setup.8'9
R% NA2 Pd or Ni Precatalyst
R + HN(R1)R2 Ligand, Base
Aryl Electrophile Amine Nucleophile N-Aryl Amino
X = Br, CI, I, OTf, OMs, OSO2NMe2
Precatalyst + Ligand Precatalyst + Ligand
ArN(R1)R2 ArX ArN(R')R2 HN(R1
)R2
VII LnM(0) VII LnNi(I)X Amine
Reductive Oxidative Reductive Ia Binding
Elimination M = Ni, Pd Addition Elimination
L,(Ar)M(11)N(R1)R2 Ln(Ar)M(II)X L,(Ar)(N(R1)R2)Ni(111)X IaLnNi(I)X Taseaai
Ha + Transmetalatic
IV II IVa HN(R1)R2
M+A-M+X-+ HA HN(R1)R2 Oxidative
* Addition 111a Amine
Amine L(Ar)M(II)X Amine ArX LnNi(I)N(R1)R2 Deprotonation
Deprotonation M+A- HN(R1)R2 Binding M+X-+ HA
Transmetalation
M(0)/M(l) Cycle M(l)/M(l) Cycle
Figure 2. Proposed catalytic cycles for Ni- and Pd-catalyzed C-N cross-coupling.
Following catalyst activation and the generation of the LnM(O) (I, Figure 2a), oxidative addition occurs with an aryl electrophile to provide the corresponding oxidative addition complex II (Figure 2). This intermediate may undergo transmetalation (amine binding and deprotonation) with the amine nucleophile to provide to corresponding amido complex (IV, Figure 2). This may then undergo reductive elimination to generate the desired aryl amine product as well as regenerate the Ni(O) or Pd(O) catalyst. The proposed M(I)/M(Ill) pathway for Ni catalysts is slightly different, with transmetalation of
LNi(I)X species Ta (Figure 2). This is then followed by oxidative addition to generate the corresponding Ni(lll) intermediate IVa (Figure 2), which can then undergo reductive elimination to provide the desired product and regenerate Ia (Figure 2). While the scope and breadth of transition metal catalyzed C-N cross-coupling has been greatly improved by recent advances in catalyst development,3 there still remains several
challenges. Here the development of new catalyst systems, particularly with regard to
ligand design, will lead to lower catalyst loadings, lower reaction temperatures, and a
broader substrate scope.
MX R = MX =MgBr, L X R -X =CI; R = EWG SNAr (Halex)
NFSI, Selectfluor Phenofluor
Addition to Deoxyfluorination Electrophilic Fluorine N2 +X-R -BE, X- = BF4, PF6, SbF6 Baiz-Schiemann 00 F R-b Aryl Fluorides Transition Metal Catalyzed or Mediated M = Ni, Pd, Cu, Ag X R-X = H, Br, OTf, SnBu3, B(OH)3 Pr Pr N N F
4
F Pr Pr F N PhO2S~ , NS 2Ph CI N+ 2BF4 -FPhenofluor NFSI Selectfluor
Figure 3. Various methods of preparing aryl C-F bonds. EWG = Electron-withdrawing
group.
OH R
As with C-N cross-coupling, methods for preparation of aryl C-F bonds has also seen many recent advances.10 Traditional methods of aryl fluoride synthesis such as the Balz-Schiemann reaction" or the Halex process" remain well-established approaches and have been frequently utilized on both small and industrial scale.1 3 1 4 However, the typical harsh reaction conditions, modest yields, and hazardous intermediates that are commonly encountered in these approaches has spurred the intense development of new methods for aryl C-F bond formation. New processes mediated or catalyzed by transition metals have been demonstrated to be effective for the fluorination for a variety of substrates.9 The dexoyfluorination of phenols15 and the quenching of aryl organometallic reagents with sources of electrophilic fluorine16 have also proved to be an effective means of accessing aryl fluorides. While these methodologies represent significant improvements, many are limited by the formation of the corresponding arene byproduct, which is often inseparable from the aryl fluoride product.17 Additionally, the substrate scope for many approaches remains limited. Therefore further development of fluorination methodologies to expand the substrate scope and suppress formation of the reduced arene is needed.
Overall, the results reported in this thesis represent the design of new transition metal catalyst systems for C-N cross-coupling and their improvements to this process. Additionally, the results reported herein detail the development of a new continuous flow process for the rapid preparation of ArF via the Balz-Schiemann, which represents a significant improvement to the standard reaction conditions for this transformation.
References
(1) (a) Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P. S. Angew. Chem.,
Int. Ed. 2011, 50, 2716. (b) Ueno, A.; Kitawaki, T.; Chida, N. Org. Lett. 2008, 10,
1999; (c) Bachovchin, W. W.; Plaut, A. G.; Drucker, D. Method of regulating
glucose metabolism, and reagents related thereto. U.S. Patent 6890898, July 3, 2002; (d) Ward, R. E.; Meyer, T. Y. Macromolecules 2003, 36, 4368; (e) Baldo,
M. A.; Lamansky, S.; Burrows, P. E.; Thompson, M. E.; Forrest, S. R. AppL. Phys.
Lett. 1999, 75, 4.
(2) (a) Gantenbein, M.; Hellstern, M.; Le Pleux, L.; Neuburger, M.; Mayor, M. Chem.
Mater. 2015, 27, 1772. (b) Roughley, S. D.; Jordan, A. M. J. Med. Chem. 2011, 54, 3451; (c) Magano, J.; Dunetz, J. R. Chem. Rev. 2011, 111, 2177; (d) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320. (3) (a) Surry, D. S.; Buchwald, S. L. Chem. Sci. 2011, 2, 27; (b) Surry, D. S.;
Buchwald, S. L. Chem. Sci. 2010, 1,13; (c) Surry, D. S.; Buchwald, S. L. Angew.
Chem. nt. Ed. 2008, 47, 6338.
(4) Tasker, S. Z.; Standley, E. A.; Jamison, T. F. Nature 2014, 509, 299.
(5) Fors, B. P.; Krattiger, P.; Strieter, E.; Buchwald, S. L. Org. Lett. 2008, 10, 3505.
(6) Ueda, S.; Su, M.; Buchwald, S. L. J. Am. Chem. Soc. 2012, 134, 700.
(7) Hie, L.; Ramgren, S. D.; Mesganaw, T.; Garg, N. K. Org. Lett. 2012, 14, 4182.
(8) (a) Standley, E. A.; Smith, S. J.; Muller, P.; Jamison, T. F. Organometallics 2014,
33, 2012; (b) Ge, S.; Green, R. A.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136,
1617; (c) Martin, A. R.; Makida, Y.; Meiries, S.; Slawin, A. M. Z.; Nolan, S. P. Organometallics 2013, 32, 6265.
(9) (a) Bruno, N. C.; Niljianskul, N.; Buchwald, S. L. J. Org. Chem. 2014, 79, 4161.
(b) Bruno, N. C.; Buchwald, S. L. Org. Lett. 2013, 15, 2876. (c) Bruno, N. C.;
Tudge, M. T.; Buchwald, S. L. Chem. Sci. 2013, 4, 916.
(10) (a) Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612; (b) Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem. Int. Ed. EngL. 2013, 52, 8214; (c)
Hollingworth, C.; Gouverneur, V. Chem. Commun. 2012, 48, 2929; (d) Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470.
(11) Balz, G; Schiemann, G. Ber. Dtsch. Chem. Ges. 1927, 60,1186.
(13) For recent examples of the use of the Balz-Schiemann reaction, see: (a) Abele,
S.; Schmidt, G.; Fleming, M. J.; Steiner, H. Org. Process Res. Dev. 2014, 18, 993; (b) Kormos, C. M.; Gichinga, M. G.; Maitra, R.; Runyon, S. P.; Thomas, J.
B.; Brieaddy, L. E.; Mascarella, S. W.; Navarro, H. A.; Carroll, F. I. J. Med. Chem. 2014, 57, 7367; (c) Hadida, S.; Van Goor, F.; Zhou, J.; Arumugam, V.; McCartney, J.; Hazlewood, A.; Decker, C.; Negulescu, P.; Grootenhuis, P. D. J.
Med. Chem. 2014, 57, 9776; (d) Gotoh, H.; Duncan, K. K.; Robertson, W. M.;
Boger, D. L. ACS Med. Chem. Lett. 2011, 2, 948; (e) Kovac, M.; Mavel, S.; Deuther-Conrad, W.; Meheux, N.; Glockner, J.; Wenzel, B.; Anderluh, M.; Brust, P.; Guilloteau, D.; Emond, P. Bioorg. Med. Chem. 2010, 18, 7659; (f) Donohue,
S. R.; Dannals, R. F. Tetrahedron Lett. 2009, 50, 7271.
(14) Moilliet, J. S. Industrial Routes of Ring-Fluorinated Aromatic Compounds. In
Organofluorine Chemistry, Principles and Commercial Applications; Banks, R. E.,
Smart, B. E., Tatlow, J. C., Eds.; Springer Science+Business Media New York: New York, 1994; pp. 195-211.
(15) Tang, P.; Wang, W.; Ritter, T. J. Am. Chem. Soc. 2011, 133,11482.
(16) For examples with aryl Grignard reagents see: (a) Yamada, S.; Gavryushin, A.; Knochel, P. Angew. Chem. Int. Ed. 2010, 49, 2215; (b) Yamada, S.; Gavryushin,
A.; Knochel, P. Angew. Chem. Int. Ed. Eng. 2010, 49, 2215.
(17) For selected examples of recent aryl fluorination methodology where the reduced arene is produced as a byproduct see: a) Ye, Y.; Schimler, S. D.; Hanley, P. S.; Sanford, M. S. J. Am. Chem. Soc. 2013, 135, 16292; b) Fier, P. S.; Luo, J.; Hartwig, J. F. J. Am. Chem. Soc. 2013, 135, 2552; c) Fier, P. S.; Hartwig, J. F. J.
Am. Chem. Soc. 2012, 134, 10795; d) Tang, P.; Furuya, T.; Ritter, T. J. Am. Chem. Soc. 2010, 132, 12150. e) Furuya, T.; Strom, A. E.; Ritter, T. J. Am. Chem. Soc. 2009, 131, 1662; f) Watson, D. A.; Su, M.; Teverovskiy, G.; Zhang,
Chapter 1
Development of an Air-Stable Nickel Precatalyst for
the Amination of Aryl Chlorides, Sulfamates,
1.1 Introduction
The development of a general, robust, and operationally simple catalyst system for Ni-catalyzed C-N cross-coupling reactions has remained a significant challenge despite of 16 years of extensive catalyst development. After our first report,' many groups have expanded Ni-catalyzed C-N cross-coupling to include new electrophiles such as aryl tosylates, carbamates, sulfamates, methyl ethers, phosphates, pivalates, and nitriles.2-4 However, many of these systems rely on air- and moisture-sensitive
Ni(COD)2 as a catalyst precursor."3 While other catalyst systems have utilized air-stable
Ni(ll) sources, many of these systems required the use of an external reductant to
generate the catalytically active Ni species.2C,5 Additionally, the overall substrate scope
of all Ni-catalyst systems reported to date has remained relatively limited, with only a few successful examples of substrates containing base-sensitive functional groups. 6-9
To address these challenges we sought to develop an air-stable, highly active Ni(II) pre-catalyst for C-N cross-coupling reactions.
Ni(Il)-(a-aryl) complexes, first reported by Shaw in 1960, were shown to be
robust compounds, stable to moisture and air.10 In 2007, Yang reported the first use of these complexes in C-N cross-coupling by demonstrating that (Ph3P)2Ni(1 -nap)CI used
in combination with an N-heterocyclic carbene ligand (lPr-HCI) generated an effective
catalyst system for the amination of aryl chlorides.'-13 Based on these precedents14 and
our previous success using dppf as a supporting ligand in Ni-based catalysis,' we
sought to investigate the use of a dppf-ligated Ni(II)-(o-aryl) complexes of as a
1.2 Results and Discussion Ph Ph dppf (Ph3P)2Ni(o-tolyl)CI T Fe Ni 1 83% -PhC. M Ph Ph 2 (dppf)Ni(o-tolyl)CI
Air-stable, yellow solid
Scheme 1. Syntheis of (dppf)Ni(o-tolyl)CI (2)
For our study, we selected (dppf)Ni(o-tolyl)CI (2) as a precatalyst for C-N bond
formation (Scheme 1). The synthesis of 2 can be accomplished by exchanging the triphenylphosphine ligands on (Ph3P)2Ni(o-tolyl)CI (1) with dppf in THF at room
temperature, resulting in an 83% yield (Scheme 1). (Ph3P)2Ni(o-tolyl)CI (1) can be easily
prepared directly from commercially available (Ph3P)2NiCI2 or in two steps from
NiCI2-6H20.15 Similar to other Ni(Il)-(o-aryl) complexes, 2a-b,5j,10,12-14 2 was found to be
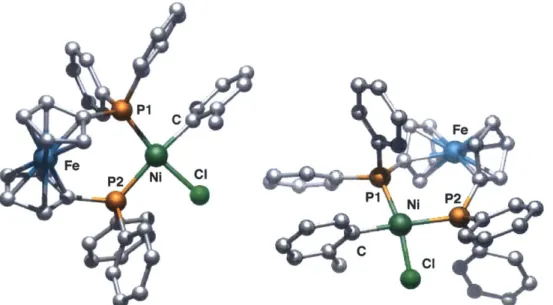
air-stable.16 A crystal structure of 2 shows a cis-ligated nickel dppf complex with square
planar geometry (Figure 1).17
P1
c
Fe
Fe P2 NI Cl
P1 Ni P2
cl
Table 1. Optimization of Reaction Conditionsa x mol % 2 0 5 mol % dppf (0) base (1.5 equiv) N N CPME (0.5 M), 100 *C H 15 min nBu nBu 3 4 5 (1.0 equiv) (1.5 equiv)
Entry mol% 2 Additive Base Conversion" Yieldb
1 5 None NaOtBu 21% 10% 2 5 None KOtBu 23% 9% 3 5 None LiOtBu 54% 49% 4 5 MeCN LiOtBu 90% 86% 5c 5 MeCN LiOtBu 100% 91% 6 5 MeCN NaOtBu 14% 5% 7 5 MeCN KOtBu 4% <4% 8d 5 MeCN LiOtBu 79% 68% 9 2.5 MeCN LiOtBu 58% 50%
10e 2.5 MeCN LiOtBu 68% 60%
aReaction conditions: 3 (0.25 mmol), 4 (0.375 mmol), base (0.375 mmol) 2 (2.5-5
mol%), dppf (2.5-5 mol%), additive (0.25 mmol), CPME (0.5 mL), 1000C, 15 minutes. bDetermined by GC using dodecane as the internal standard. cReaction time was 45
min. Isolated yield was 85%,1 mmol scale, average of two runs. dNo additional dppf was added. eReaction time was 1 h. CPME = cyclopentyl methyl ether.
Having prepared and characterized 2, we set out to investigate its use as a
precatalyst in the amination of 4-n-butylchlorobenzene (3) with morpholine (4) (Table 1). Performing the reaction using 5 mol % of 2 and 5 mol % dppf as the catalyst with
NaOtBu as the base in CPME for 15 minutes at 100 0C resulted in 10% yield (entry 1,
Table 1). KOtBu as a base provided a similar result (9% yield), while the use of LiOtBu led to a marked improvement in reactivity, giving the coupled product (5) in 49% yield (entries 2 and 3, Table 1), demonstrating that the choice of counterion is crucial to the success of the reaction when using tert-butoxide bases.8 Previously, Hartwig has shown that the addition of benzonitrile improved the reactivity in Ni-based systems-potentially stabilizing the catalytically active Ni-species.19 Similarly, we found that the addition of 1 equivalent of acetonitrile greatly improved the reactivity of 2, producing the product 5 in 86% yield (entry 4, Table 1). Extending the reaction time to 45 minutes
gave full conversion of the aryl chloride, and product 5 was isolated in 85% yield (entry
5, Table 1). The use of the acetonitrile additive with either NaOtBu or KOtBu, however,
still resulted in low yields (entry 6 and 7, Table 1). Conducting the reaction without an additional equivalent of the dppf ligand led to a diminished yield of 5 (entry 8, Table 1), and lowering the catalyst loading had a similarly detrimental effect, with the reaction failing to reach full conversion even after extended reaction times (entries 9 and 10,
Table 1).
In addition to the desired arylamine product 5, a small amount of the corresponding aminated o-tolyl product (2%) was also observed. This byproduct is likely the result of catalyst activation, and similar byproducts have been observed in other
systems using Ni(Il)-(o-aryl) complexes as precatalysts.20 We note that the
reactions using 2. In the instances when they were formed, the small amounts (0.5
-5%) were easily separated during purification.2 2
Table 2. Amination of Aryl Chlorides using LiOtBua 5 mol % 2, 5 mol % dppf MeCN, LiOtBu ArCi + R2NH IN CPME, 100 'C, 1 h Ar-NR2 K N --*Me (N)
KNI
N 6ab 78% & N0 N N MeO OMe 6dc 82% 0 Me 6b 83% N 6e 60% 3FCN N 6f 98%aReaction conditions: aryl chloride (1.0 mmol), amine (1.5 mmol), LiOtBu (1.5 mmol), 2
(5 mol%), dppf (5 mol%), MeCN (1.0 mmol), CPME (2 mL), 100 C, 1 h. Yields are of the
isolated product, average of two runs. bReaction time 16 h. c2 (10 mol%) and dppf (10
mol%), 1300C, 16 h.
With the conditions for amination identified, we investigated the scope of this
reaction with aryl and heteroaryl chlorides. The catalyst system was found to tolerate
6c
both cyclic and acyclic secondary alkylamines, which were coupled in high yield (6a and
6b, Scheme 2). Primary anilines, including the bulky, ortho-substituted 2-aminobiphenyl,
were readily arylated in excellent yield (6c and 6f, Scheme 2). Diphenylamine also underwent facile cross-coupling with 2-chloro-4,6-dimethoxypyrimidine in 82% yield, although the reaction required increased catalyst loading and higher temperatures to achieve full conversion (6d, Scheme 2). The arylation of indoline with 1-chloronaphthalene suffered from significant reduction of the aryl halide presumably due
to competing p-hydride elimination, but the desired product was still isolated in 60% yield (6e, Scheme 2).
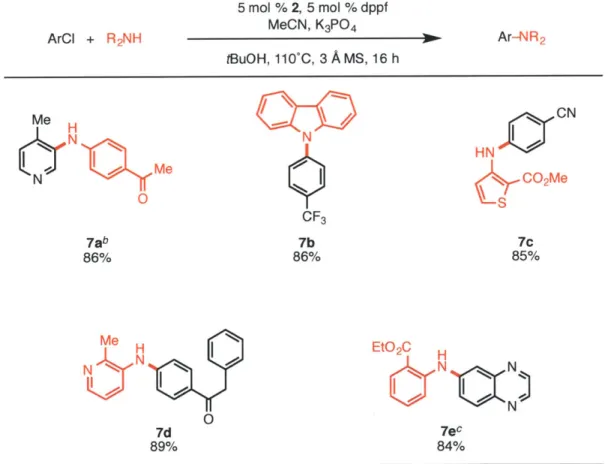
Table 3. Amination of Aryl Chlorides using K3PO4a
5 mol % 2, 5 mol % dppf
ArCI + R2NH MeCN, K3PO4 3 Ar-NR2
tBuOH, 11OC, 3 A Ms, 16 h Me H CN CF3 7ab 7b 7c 86% 86% 85% Me H EtO2C N N N ON 7d 7 89% 84%
aReaction conditions: aryl chloride (1.0 mmol), amine (1.5 mmol), K3PO4 (3 mmol), 2 (5
mol%), dppf (5 mol%), MeCN (1.0 mmol), tBuOH (2 mL), 3A MS (300 mg), 110 C, 16 h.
Yields are of the isolated product, average of two runs. bIn cases where using the standard conditions do not give full conversion, omission of the 3A MS or using 6 mmol of K3PO4 were found to allow the reaction to reach completion. CK3PO4 (6 mmol),
Dioxane (4 mL).
Although our catalyst system proved to be highly effective for amination of aryl and heteroaryl chlorides using LiOtBu, we were interested in expanding the scope of the reaction to include substrates bearing base sensitive functional groups. Due to their lower pKa values relative to alkylamines, we felt that anilines would be readily
deprotonated during the transmetallation step (amine binding and deprotonation) under weakly basic conditions.23 Thus, by changing the solvent to tBuOH and employing
K3PO4 as the base,
2 4 we found that a wide variety of primary and secondary anilines
could be efficiently arylated (Scheme 3). These conditions were found to tolerate base-sensitive functional groups as well as ortho-substitution on the aryl halide and the amine nucleophiles, including anilines containing an ester substituent in the ortho position (Scheme 3).
Having established the conditions for the arylation of anilines using K3PO4, we
sought to expand the reaction scope further to include phenol-derived aryl electrophiles.
While the amination of aryl sulfamate electrophiles is well-established, the amination of aryl mesylates with Ni-catalyst systems remains unknown.25,26
has been reported.27 As aryl mesylate and some aryl triflate electrophiles are known to
be sensitive to strong bases, 24a,2 we felt that application of the developed weak base
conditions would enable the successful amination of these electrophiles. However, the use of tBuOH proved to be detrimental to the reaction, resulting in modest yields of the desired product. By using CPME as the solvent and performing the reaction at a slightly lower- concentration a dramatic improvement of the product yields was observed. As with the aryl chloride substrates, these conditions were able to tolerate base sensitive functional groups as well as ortho-substituents on either the electrophile or the nucleophile (Scheme 4).
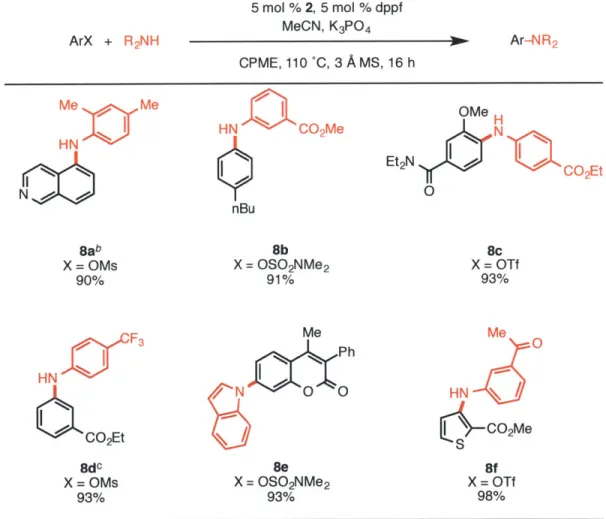
Table 4. Amination of Aryl Mesylates, Triflates, and Sulfamatesa
ArX + R2NH Me Me N Bab X = OMs 90% HN F3 CO2Et 8dc X = OMs 93% 5 mol % 2, 5 mol % d MeCN, K3PO4 CPME, 110 'C, 3 A Ms, HNQaCo2Me nBu 8b X = OS0 2NMe2 91% Me / Ph N 0 0 8e X = OSO2NMe2 93% ppf 16 h Ar-NR2 OMe H Et2N CO2Et 0 8c X = OTf 93% Me 0 HN S\ - C02Me 8f X = OTf 98%
aReaction conditions: aryl chloride (1.0 mmol), amine (1.5 mmol), K3PO4 (3 mmol), 2 (5
mol%), dppf (5 mol%), MeCN (1.0 mmol), CPME (4 mL), 3A MS (300 mg), 110 C, 16 h.
Yields are of the isolated product, average of two runs. In cases where using the standard conditions do not give full conversion, using 6 mmol of K3PO4 were found to
allow the reaction to reach completion. c2 (2.5 mol%), dppf (2.5 mol), 4 h.
1.3 Conclusions
In summary, we have developed a highly active, dppf-ligated nickel precatalyst (2) for use in C-N cross-coupling reactions. This robust precatalyst can be easily
prepared from readily available Ni(II) sources and is air-stable. Furthermore, this catalyst system has been demonstrated to cross-couple a wide array of amine nucleophiles efficiently with aryl and heteroaryl electrophiles, including substrates containing base sensitive functional groups.
1.4 Experimental
General Experimental Information: All reactions were set up on the bench top
and run under a nitrogen atmosphere unless otherwise noted. All reagents and precatalyst (2) were weighed out in air. HPLC grade THF was purchased from Macron and purged with argon for 30 minutes and then passed through two packed columns of
neutral alumina under argon pressure. HPLC grade dichloromethane was purchased from J. T. Baker and was passed through two packed columns of neutral alumina under
tert-butanol (tBuOH) were purchased from Aldrich in Sure-Seal@ bottles and used as received. Lithium tert-butoxide, sodium tert-butoxide, and potassium tert-butoxide were
purchased from Aldrich and potassium phosphate was purchased from Acros. Sodium tert-butoxide, potassium tert-butoxide, and potassium phosphate were stored in a nitrogen-filled glovebox and small amounts (3-4 g) were removed and stored in air in a desiccator filled with calcium sulfate. Lithium tert-butoxide was stored in air in a desiccator filled with calcium sulfate. Carbazole was purchased from Aldrich and
recrystallized from toluene. 1,1 '-bis(diphenylphosphene)ferrocene (dppf) and
bis(triphenylphosphine)nickel(11) dichloride were purchased from Strem and used as received. Both (Ph3P)2Ni(o-tolyl)CI (1) and (dppf)Ni(o-tolyl)CI (2) were stored at room
temperature under air in a desiccator filled with calcium sulfate. All other commercially available reagents were used as received without further purification unless otherwise noted.
General Analytical Information: All compounds were characterized by 1H, 13C, IR, and either elemental analysis or high-resolution mass spectrometry (HRMS). The 1H and 13C spectra are available in section 1.6 following the references. NMR analyses were performed on Varian 500 MHz, Varian 300 MHz or Bruker 400MHz instruments. Chemical shifts for 1H NMR were measured relative to the residual solvent signals of
C6D6 (7.16 ppm), CDCI3 (7.26 ppm), CD2CI2 (5.32 ppm) and CD3OD (3.31 ppm).
Chemical shifts for 13C NMR were measured relative to the residual solvent signals of
C6D6 (128.06 ppm), CDCI3 (77.16 ppm), and CD2CI2 (54.00 ppm). All 19F chemical shifts
31P NMR chemical shifts are in 8 (ppm) units relative to an external standard H
3PO4
(0.00 ppm).
Yields refer to the isolated yield of compounds with greater than 95% purity as determined by gas chromatography (GC), 1H NMR, 13C NMR, and elemental analysis. The GC yields and GC conversions in Table 1 were determined from the crude reaction mixture using dodecane as the internal standard. GC analyses were performed on an Agilent 6890 gas chromatograph using a J&W DB-1 column with a FID detector. The infrared (IR) data collected were of the neat compound and were collected using instrument Thermo Scientific - Nicolet iS5 (iD5 ATR - Diamond). Elemental analysis of
samples was performed by Atlantic Microlabs Inc., Norcross, GA. Flash
chromatography was performed using SiliaFlash F60 silica gel from SiliCycle. In some cases, flash chromatography was performed with the aid of a Biotage SP4 instrument
using silica-packed cartridges.
1.4.1 Synthesis of Starting Materials
Preparation of Aryl Mesylates: All aryl mesylates were known and synthesized
according to literature procedures.30
Preparation of Aryl Sulfamates:
Me To a 300 mL round bottom flask equipped with a Ph magnetic stir-bar, was added
7-hydroxy-4-methyl-3-Me2NO2SO phenylcoumarin (2.61 g, 10.4 mmol). The flask was
fitted with a septum and purged with nitrogen. Dichloromethane (15 mL) and DBU (3.10 mL, 20.7 mmol) were then added via syringe followed by dropwise addition of
NN-dimethylsulfamoyl chloride (1.17 mL, 10.9 mmol) via syringe. The reaction mixture was then allowed to stir at room temperature for 3 h. After 3 h, the reaction mixture was poured into water (50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated with the aid
of a rotary evaporator. The crude product was purified via Biotage SP4 (silica-packed
100 g Snap cartridge; using a 0-50% ethyl acetate in hexanes gradient) to provide the
title compound as a white solid (1.80 g, 49%), mp = 152 - 154 *C.
1H NMR (500 MHz, CDC1 3) 6 7.70 - 7.68 (m, 1 H), 7.46 - 7.43 (m, 2H), 7.40 - 7.37 (m, 1 H), 7.32 - 7.23 (m, 4H), 3.01 (s, 6H), 2.30 (s, 3H). 13C NMR (126 MHz, CDC13) 6 160.48, 153.17, 152.03, 147.18, 134.06, 129.97, 128.44, 128.34, 127.05, 126.55, 119.09, 117.86, 109.96, 38.79, 16.75. FTIR (neat, cm-1): 3071.35, 2983.97, 1724.43, 1608.22, 1567.59, 1496.29, 1423.68, 1363.52, 1251.2, 1178.97, 1147.6, 1124.04, 1070.79, 1036.89, 1003.17, 987.62, 954.53, 905.26, 830.9, 813.69, 783.13, 732.02, 721.42, 700.41, 669.39, 643.79, 610.01, 557.38, 538.1.
4-butylphenyl dimethylsulfamate (10)
OSO2NMe2 An oven-dried round bottom flask was equipped with a magnetic
stir-bar and sodium hydride (60% dispersion in mineral oil, 583 mg, 14.6 mmol) was added. After fitting with a septum and purging with
nBu
nitrogen, THF (35 mL) was added and the reaction mixture was cooled to 0 0C in an ice
bath. A solution of 4-n-butylphenol (2.1 g, 13.9 mmol) in THF (5 mL) was then added dropwise via syringe (the septum was punctured with a second needle to vent the evolved gases) and stirred at 00C for 30 min. After 30 min, a solution of NN-dimethylsulfamoyl chloride (2.09 g, 14.6 mmol) in THF (5 mL) was then added dropwise via syringe. The reaction mixture was then warmed to room temperature and stirred for 24 h. After 24 h, the reaction mixture was quenched with H20, and extracted with Et20
and the combined organic layers were washed with aqueous 1 M KOH. The organic layers were then dried over Na2SO4, filtered and concentrated with the aid of a rotary
evaporator. The crude residue was then purified by flash chromatography (50% CH2C2
in hexane) to give the title compound as a colorless oil (2.0 g, 56%).
1H NMR (500 MHz, C6D 6) 6 7.20 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 2.49 (s, 6H), 2.39 - 2.30 (m, 2H), 1.40 - 1.33 (m, 2H), 1.21 -1.13 (m, 2H), 0.82 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, C6D6) 6 148.94, 141.46, 129.81, 121.99, 38.31, 35.19, 33.78, 22.56, 14.11.
FTIR (neat, cm-1): 2956.04, 2929.77, 2858.18, 1502.13, 1458.83, 1414.71, 1367.91,
1275.69, 1195.52, 1171.66, 1148.42, 1055.24, 969.83, 857.29, 806.4, 784.39, 746.22, 682, 637.66.
Anal. Calcd. for C12H19NO3S: C, 56.01; H, 7.44. Found: C, 56.24; H, 7.65.
Preparation of Aryl Triflates: 4-(diethylcarbamoyl)-2-methoxypheny trifluoromethanesulfonate was prepared according to the literature procedure.31
Methyl 3-(((trif I uoromethyl)sulfonyl)oxy)thiophene-2-carboxylate (11)
OTf To a flame dried 300 mL round bottom flask,
3-hydroxythiophene-2-CO2Me carboxcylic acid methyl ester (949 mg, 6.0 mmol) was added and the
S flask was fitted with a septum and purged with nitrogen.
Dichloromethane (12 mL) and triethylamine (1.67 mL, 12 mmol) were then added and
the reaction flask was cooled to -78 CC in a dry ice/acetone bath.
Trifluoromethanesulfonic anhydride (1.21 mL, 7.2 mmol) was then added dropwise via syringe and the reaction mixture was stirred at -78 OC for 1 h. The cold bath was then removed and the reaction mixture was allowed to warm to room temperature and stirred
at room temperature for an additional 1 h. The reaction mixture was poured onto water
(50 mL) and extracted with dichloromethane (3 x 50 mL). The combined organic layers
were dried over Na2SO4, filtered, and concentrated with the aid of a rotary evaporator.
The crude product was purified via flash chromatography (5% Et20 in hexanes) to
'H NMR (500 MHz, C6D6) 6 6.71 (d, J= 5.5 Hz, 1H), 6.50 (d, J= 5.5 Hz, 1H), 3.43 (s, 3H). 13C NMR (126 MHz, C6D6) 6 159.87, 145.64, 130.68, 123.13 (q, J= 320.7 Hz), 122.77, 122.10, 52.03. 19F NMR (282 MHz, C 6D6) 6 -74.41. FTIR (neat, cm-1): 3122.3, 2958.21, 1719.67, 1539.6, 1423.07, 1385.92, 1298.48, 1268.6, 1248.93, 1205.94, 1135.75, 1087.8, 1015.77, 1003.32, 941.59, 884.25, 855.15, 814.22, 773.85, 763.56, 656.1, 601.18.
HRMS-ESI (m/z) Calcd for C7H5F305S2 [M+Na]: 312.9423; Found: 312.9419.
1.4.2 Preparation of Nickel Precatalyst
[Bis(triphenylphosphine)](o-tolyl)chloronickel (1)
Me Bis(triphenylphosphine)nickel(II) dichloride (11.4 g, 17.4 mmol) was added to an oven-dried 500 mL round bottom flask equipped with a
Ni -Cl
PPh3 magnetic stir-bar and purged with nitrogen. THF (240 mL) was added
via syringe and the reaction mixture was cooled to 0 0C in an ice bath. o-tolylmagnesium
chloride (22.8 mL, 0.8 M in THF, 18.3 mmol) was then added dropwise via syringe. The reaction mixture was stirred for 15 min at 00C and an additional 1 h at room temperature
and then was quenched with methanol (15 mL). The mixture was concentrated with the aid of a rotary evaporator and the resulting residue was triturated with the aid of sonication in methanol (450 mL) to produce a yellow suspension. The yellow
suspension was filtered and the yellow solid was washed with methanol. The solid was then dissolved in CH2C2 (80 mL) and carefully filtered through a Whatman GF/D glass
fiber filter to afford a homogenous orange solution that was concentrated with the aid of a rotary evaporator. The resulting orange solid was collected and triturated with pentane
to afford the title compound 1 as a yellow solid (7.4 g, 60%) mp = 166 0C
(decomposition). 1H NMR (500 MHz, CD 2C2) 6 7.51 - 7.11 (m, 31 H), 6.34 - 6.27 (m, 2H), 5.95 (s, 1 H), 2.09 (s, 3H).) 13C NMR (126 MHz, CD2C 2) 6 144.02, 136.17, 135.15 (t, J = 5.1 Hz), 132.28 (t, J= 21.4 Hz), 130.09, 129.59, 128.22 (t, J= 4.3 Hz), 123.15, 122.57, 26.46. 31P NMR (121 MHz, CD 2C2) 621.72. FTIR (neat, cm-1): 3052.84, 1568.63, 1480.03, 1431.66, 1304.72, 1184.77, 1155.73, 1093.46, 1027.95, 852.78, 737.84, 689.06.
Anal. Calcd. for C43H37CINiP2: C, 72.76, H, 5.25. Found: C, 72.66, H, 5.39.
[1,1'-Bis(diphenylphosphino)ferrocene](o-tolyl)chloronickel (2)
[Bis(triphenylphosphine)](o-tolyl)chloronicke (1) (2.95 g, 4.17 Ph Ph
mmol) and 1,1'-Bis(diphenylphosphino)ferrocene (2.66 g, 4.80
Fe NiP
Fe Ni Me mmol) were added to an oven-dried 500 mL round bottom flask PCl
Ph Ph equipped with a magnetic stir-bar and stoppered with a rubber septum. The sealed flask was evacuated and refilled with nitrogen (this sequence was repeated a total of three times) and the THF (64 mL) was added via syringe. The orange
mixture was allowed to stir at room temperature for 2 h. Pentane (144 mL) was then
added via syringe and the resulting mixture was allowed to stir for 2 h to produce a yellow suspension. Note: Addition of the correct amount of pentane is critical to facilitate the completion of the ligand exchange. Additional pentane (100 mL) was added and the
product was isolated by filtration and washed with pentane to produce an orange solid. The solid was dissolved in dichloromethane (10 mL), and the resulting red suspension was carefully filtered through a Whatman GF/D glass fiber filter fitted inside a fritted filter (medium frit) to collect a homogenous red solution in a 300 mL round bottom flask that was then concentrated with the aid of a rotary evaporator (bath temperature was set to
250C). The resulting residue was then triturated in a large excess of pentane with the
aid of sonication to produce a yellow suspension. Note: The filtration and trituration are
crucial steps for obtaining a highly active precatalyst. However, this complex will decompose in a dichloromethane solution after an extended period of time and therefore the filtration and trituration steps must be done promptly after addition of dichloromethane. The solid was isolated by filtration to collect the title compound as a yellow solid (2.7 g, 83%) mp = 166 - 167 0C (decomposition).
1H NMR (500 MHz, CD2C2) 6 8.28 (s, 4H), 8.08 (s, 2H), 7.54 (m, 7H), 7.36 (s, 2H), 7.29
(s, 1H), 7.09 (s, 1H), 6.86 (s, 2H), 6.70 (s, 2H), 6.53 (s, 1H), 6.40 (s, 1H), 6.20 (s, 1H),
5.25 (s, 1H), 4.64 (s,1H) 4.34 (s, 1H), 4.28 (s, 1H), 4.13 - 4.11 (m, 2H), 3.62 (s, 1H), 3.42 (s, 1 H), 2.54 (s, 3H).
13C NMR (126 MHz, CD2C2) Complex spectrum, see attached.
FTIR (neat, cm-'): 3046.18, 1568.59, 1481.79, 1431.62, 1305.35, 1190.43, 1162.86,
1096.72, 1037.59, 1009.94, 814.67, 733.75, 696.39, 688.23, 640.69.
Anal. Calcd. for C41 H35CIFeNiP2: C, 66.58, H, 4.77. Found: C, 66.34, H, 4.85.
Crystals suitable for X-ray diffraction were obtained by slow vapor diffusion of pentane onto a THF/CH2Cl2 solution of the complex at room temperature.
1.4.3 Procedures for the Nickel-catalyzed C-N Cross-coupling
General Procedure for Table 1. To an oven-dried disposable test tube (Fisher
Scientific, catalog number: 1495925C) equipped with a screw-cap septum and a magnetic stir-bar (dppf)Ni(o-tolyl)CI (2) (9.2 mg, 0.0125 mmol), dppf (6.9 mg, 0.0125 mmol), and base (0.375 mmol) were added. The reaction vessel was evacuated and backfilled with nitrogen. This sequence was repeated a total of three times.
4-n-butylchlorobenzene (0.042 mL, 0.25 mmol) and morpholine (0.032 mL, 0.375 mmol) were then added via syringe. Any additive, if liquid, was also added at this time. CPME
(0.5 mL) was then added and the screw-cap septum was exchanged for one with an
unpunctured septum under a continuous flow of nitrogen. The reaction mixture was then heated to 100 0C in a pre-heated oil bath for 15 min. After cooling to room temperature,
dodecane (0.057 mL, 0.25 mmol) was added and the reaction mixture was diluted with ethyl acetate. A small aliquot was filtered through a short silica gel plug and analyzed by GC.
General Procedure A. To an oven-dried disposable test tube (Fisher Scientific,
catalog number: 1495937A) equipped with a screw-cap septum and a magnetic stir-bar
(dppf)Ni(o-tolyl)CI (2) (0.05 mmol), dppf (0.05 mmol), and LiOtBu (1.5 mmol) were added. Aryl halide (1 mmol) and amine (1.5 mmol), if solid, were also added at this point. The reaction vessel was then evacuated and backfilled with nitrogen. This
sequence of evacuation and backfilling with nitrogen was repeated a total of three times.
Aryl halide and amine, if liquid, were added, followed by MeCN (0.052 mL, 1 mmol) and CPME (2 mL). The cap was replaced with one containing an unpunctured septum under a continuous flow of nitrogen and the reaction mixture was heated to 100 0C for the
specified time in a pre-heated oil bath. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate and filtered through Celite eluting with additional ethyl acetate. The filtrate was then concentrated with the aid of a rotary evaporator and the residue was purified via flash chromatography.
4-(4-butylphenyl)morpholine (5)
O Following general procedure A, a mixture of 4-n-butylchlorobenzene (0.169 mL,
N 1 mmol), morpholine (0.129 mL, 1.5 mmol), LiOtBu (120 mg, 1.5 mmol),
(dppf)Ni(o-tolyl)CI (2) (37 mg, 0.05 mmol), dppf (28 mg, 0.05 mmol), MeCN
oO (0.052 mL, 1 mmol) and CPME (2 mL) was heated to 100 0C for 45 minutes.
(silica-packed 50 g Snap cartridge, 0-5% ethyl acetate in hexanes) to provide the title
compound as a orange solid (Run 1: 186 mg, (85%); Run 2: 185 mg (84%); Average Yield: 85%) mp = 37-39 *C. 1H NMR (500 MHz, C 6D6) 6 7.06 (d, J = 8.4 Hz, 2H), 6.70 (d, J = 8.4 Hz, 2H), 3.59 3.57 (m, 4H), 2.78 2.76 (m, 4H), 2.52 (t, J = 7.7 Hz, 2H), 1.59 1.52 (m, 2H), 1.34 -1.32 (m, 2H), 0.89 (t, J= 7.4 Hz, 3H). 13C NMR (126 MHz, C 6D6) 6 150.09, 134.31, 129.28, 116.33, 67.03, 50.01, 35.21, 34.34, 22.69, 14.25. FTIR (neat, cm'): 2957.6, 2921.49, 2858.94, 2829.22, 1610.39, 1514.1, 1444.81, 1379.73, 1363.95, 1327.79, 1301.41, 1261.04, 1230.42, 1119.59, 1068.7, 860.21, 840.53, 809.07, 730.68, 704.79, 627.09.
Anal. Calcd. for C14H21NO: C, 76.67, H, 9.65. Found: C, 76.78, H, 9,51. Characterization
data are in accordance with those in the literature.
2-methyl-8-(4-(pyridin-2-yl)piperazin-1-yl)quinoline (6a)
Following general procedure A, a mixture of
8-chloro-2-N Me methylquinoline (178 mg, 1.0 mmol), 1-(2-pyridyl)piperazine (0.228
N mL, 1.5 mmol), LiOtBu (120 mg, 1.5 mmol), (dppf)Ni(o-tolyl)CI (2) N (37 mg, 0.05 mmol), dppf (28 mg, 0.05 mmol), MeCN (0.052 mL,
N 1.0 mmol) and CPME (2 mL) was heated to 100 0C for 16 h. The
reaction mixture was concentrated with the aid of a rotary
acetate in hexanes with 1% triethylamine) to provide the title compound as a yellow solid (Run 1: 232 mg, (76%); Run 2: 241 mg, (79%) Average Yield: 78%), mp =
123-126 *C. 1H NMR (500 MHz, CDC1 3) 6 8.25-8.24 (m, 1 H), 7.99 (d, J = 8.5 Hz, 1 H), 7.53-7.49 (m, 1H), 7.43-7.35 (m, 2H), 7.26 (d, J= 8.4 Hz, 1H), 7.13 (d, J= 6.8 Hz, 1H), 6.75 (d, J= 8.6 Hz, 1 H), 6.70-6.60 (m, 1 H), 3.89-3.87 (m, 4H), 3.56-3.54 (m, 4H), 2.75 (s, 3H). 13C NMR (126 MHz, CDC13) 6 159.86, 156.94, 148.54, 148.03, 141.99, 137.59, 136.71, 129.74, 127.79, 125.81, 121.70, 115.96, 113.41, 107.30, 51.98, 45.58, 25.97. FTIR (neat, cm1): 3000.59, 2971.61, 2846.94, 2816.98, 1592.1, 1561.28, 1481.15, 1431.86, 1383.63, 1369.87, 1352.67, 1310.8, 1232.49, 1154.39, 1118.17, 1087.67, 1024.57, 979.78, 943.62, 843.5, 772, 756.39, 737.16, 711.97, 696.6. Anal. Calcd. for C19H20N4: C, 74.97; H, 6.62. Found: C, 74.91; H, 6.58.
2-(methyl(phenethyl)amino)-9H-thioxanthen-9-one (6b)
0 Me Following general procedure A, a mixture of
2-chlorothioxanthone (247 mg, 1.0 mmol),
N-imethylphenethylamine (0.218 mL, 1.5 mmol),
LiOtBu (120 mg, 1.5 mmol), (dppf)Ni(o-tolyl)CI (2) (37 mg, 0.05 mmol), dppf (28 mg, 0.05 mmol), MeCN (0.052 mL, 1.0 mmol) and CPME (2 mL) was heated to 100 0C for 1 h. Purification via flash chromatography (0 - 10% ethyl acetate in hexanes) provided the title compound as a yellow solid (Run 1: 286 mg, (83%); Run 2: 286 mg, (83%) Average Yield: 83%), mp = 102-103 0C.
'H NMR (500 MHz, CDC13) 6 8.65 (d, J= 8.1 Hz, 1H), 7.91 (d, J= 3.0 Hz, 1H), 7.56 (m, 2H), 7.51-7.38 (m, 2H), 7.33-7.30 (m, 2H), 7.24-7.20 (m, 3H), 7.09 (d, J= 7.1 Hz, 1H), 3.67 (m, 2H), 2.97 (s, 3H), 2.90 (m, 2H). 13C NMR (126 MHz, CDCI3) 6 180.10, 147.52, 139.41, 137.93, 131.70, 130.09, 129.92, 128.94, 128.80, 128.69, 126.99, 126.42, 126.08, 125.65, 123.78, 118.53, 110.39, 54.73, 38.81, 32.97. FTIR (neat, cm-1): 2934.14, 1626.82, 1592.04, 1493.5, 1450.64, 1433.21, 1413.04, 1383.19, 1351.28, 1332.64, 1219.48, 1193.04, 1135.51, 1116.9, 1073.79, 1033.12, 942.57, 859.05, 800.32, 737.92, 697.78.
HRMS-ESI (m/z) Calcd. for C22H19NOS [M+H]: 346.1260; Found: 346.1249.
N-([,1 '-biphenyl]-2-yl)pyridin-3-amine (6c)
Following general procedure A, a mixture of 3-chloropyridine (0.095 mL, 1.0 mmol), 2-aminobiphenyl (254 mg, 1.5 mmol), LiOtBu (120
H
N mg, 1.5 mmol), (dppf)Ni(o-tolyl)CI (2) (37 mg, 0.05 mmol), dppf (28
N /! mg, 0.05 mmol), MeCN (0.052 mL, 1 mmol) and CPME (2 mL) was
heated to 100 0C for 1 h. Purification via flash chromatography (40% ethyl acetate in hexanes) provided the title compound as an off-white solid (Run 1: 233 mg, (94%); Run 2: 234 mg, (94%); Average Yield: 94%), mp = 132-134 *C.
1H NMR (500 MHz, CDC1
3) 8 8.28 (s, 1H), 8.15 (d, J = 1 Hz, 1H), 7.45-7.43 (m, 4H), 7.38-7.34 (m, 3H), 7.31-7.28 (m, 2H), 7.16-7.14 (m, 1H), 7.09-7.06 (m, 1H), 5.70 (s,
13C NMR (126 MHz, CDC13) 6 141.97, 140.45, 140.20, 139.02, 138.75, 132.59, 131.22, 129.29, 129.03, 128.46, 127.74, 123.77, 123.73, 122.35, 118.15.
FTIR (neat, cm~1): 3233.66, 2922, 1575.26, 1516.02, 1469.64, 1432.68, 1407.25,
1345.86, 1302.82, 1161.49, 1103.72, 1046.64, 1020.88, 1008.39, 884.83, 789.27, 778.47, 756.51, 742.69.
Anal. Calcd. for C17H14N2: C, 82.90; H, 5.73. Found: C, 82.46; H, 5.91.
4,6-dimethoxy-N, N-diphenyl pyrimidi n-2-amine (6d)
Following general procedure A, a mixture of
2-chloro-4,6-dimethoxypyrimidine (174 mg, 1 mmol),
diphenylamine (254 mg,
1.5 mmol), LiOtBu (120 mg, 1.5 mmol), (dppf)Ni(o-tolyl)CI (2) (74
N N
Il
mg, 0.05 mmol), dppf (55 mg, 0.05 mmol), MeCN (0.052 mL, 1MeO OMe
mmol) and CPME (2 mL) was heated to 130 0C for 16 h. The
reaction mixture was filtered through a plug of Celite and eluted with additional ethyl
acetate. The crude product was adsorbed to silica gel and purified via Biotage SP4
(silica-packed 50 g Snap cartridge, 0 - 50% CH2CI2 in hexanes) to provide the title
compound as a brown solid (Run 1: 242 mg, (78%); Run 2: 264 mg, (86%); Average Yield: 82%), mp = 154-155 *C. 1H NMR (500 MHz, CD 2CI2) 8 7.43-7.40 (m, 8H), 7.27-7.24 (m, 2H), 5.64 (s, 1H), 3.72 (s, 6H). 13C NMR (126 MHz, CD2C 2) 6 172.21, 161.22, 145.28, 129.09, 128.33, 125.69, 81.42, 54.0.
FTIR (neat, cm-1): 2948.28, 1566.38, 1491.72, 1462.15, 1423.02, 1398.31, 1362.05,
1286.48, 1241.36, 1191.31, 1156.54,1059.79, 1000.54, 952.44, 798.79, 758.13.
Anal. Calcd. for C18H17N302: C, 70.34; H, 5.58. Found: C, 70.24; H, 5.63.
1-(naphthalen-1-yI)indoline (6e)
Following general procedure A, a mixture of 1-chloronapthalene (0.136
Q 2N
mL, 1 mmol), indoline (0.168 mL, 1.5 mmol), LiOtBu (120 mg, 1.5 mmol), (dppf)Ni(o-tolyl)CI (2) (37 mg, 0.05 mmol), dppf (28 mg, 0.05 mmol), MeCN (0.052 mL, 1 mmol) and CPME (2 mL) was heated to 100 0C for 1h. The reaction mixture was then cooled to room temperature and diluted with Et20 and
filtered through a plug of silica gel, eluting with excess Et20. The filtrate was
concentrated with the aid of a rotary evaporator and the crude residue was purified via flash chromatography (pentane) to provide the title compound as a white solid (Run 1:
148 mg, (60%); Run 2:148 mg, (60%); Average Yield: 60%), mp = 80-83 *C.
1H NMR (500 MHz, C 6D6) 6 8.21 (d, J= 7.5 Hz, 1H), 7.69 (d, J= 7.7 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.30-7.23 (m, 3H), 7.19 (d, J= 7.4 Hz, 1H), 7.10 (d, J= 7.2 Hz, 1H), 6.88 (t, J= 7.6 Hz, 1H), 6.76 (t, J= 7.3 Hz, 1H), 6.32 (d, J= 7.9 Hz, 1H), 3.54 (m, 2H), 2.80 (m, 2H). 13C NMR (126 MHz, C6D6) 6 151.54, 142.95, 135.36, 131.05, 130.68, 128.70, 127.56, 126.51, 126.41, 126.00, 125.79, 124.90, 124.76, 119.95, 118.99, 109.67, 55.63, 29.19. FTIR (neat, cm-1): 3051.94, 3026.43, 2978.58, 2947.66, 2892.19, 2867.49, 2842.32, 1604.86, 1575.09, 1482.89, 1456.1, 1398.47, 1329.65, 1297.25, 1262.55, 1222.81,
1163.19, 1117.51, 1073.03, 1050.31, 1026.62, 973.63, 947.57, 925.25, 872.16, 799.37,
765.44, 749.14, 736.11, 648.37, 641.83.
Anal. Calcd. for C18H15N: C, 88.13; H, 6.16. Found: C, 88.28; H, 6.07.
N-(3-(trifluoromethyl)phenyl)quinolin-6-amine (6f)
H Following general procedure A, a mixture of
6-F3CN
z
*
chloroquinoline (164 mg, 1 mmol),
3-N trifluoromethylaniline (0.187 mL, 1.5 mmol), LiOtBu (120
mg, 1.5 mmol), (dppf)Ni(o-tolyl)Cl (2) (37 mg, 0.05 mmol), dppf (28 mg, 0.05 mmol), MeCN (0.052 mL, 1 mmol) and CPME (2 mL) was heated to 100 0C for 1 h. Purification
via flash chromatography (20-40% ethyl acetate in hexanes) provided the title compound as a white solid (Run 1: 278 mg, (97%); Run 2: 285 mg, (99%); Average Yield: 98%), mp = 170-172 *C. 1H NMR (500 MHz, CDC1 3) 6 8.76 (s, 1H), 8.05 (d, J= 9.0 Hz, 1H), 8.00 (d, J= 8.3 Hz, 1 H), 7.50-7.31 (m, 5H), 7.23 (d, J= 7.3 Hz, 1H), 6.33 (s, 1H). 13C NMR (126 MHz, CDC13) 6 148.24, 144.71, 143.19, 140.59, 134.91, 132.21 (q, J= 32.3 Hz), 130.92, 130.17, 129.59, 125.19 (q, J = 272.6 Hz), 123.64, 121.83, 121.11, 118.31 (q, J= 3.3 Hz), 114.78 (q, J= 3.8 Hz), 111.37. 19F NMR (471 MHz, CDC 3) 6 -63.06. FTIR (neat, cm'): 3249, 3042.68, 2941.26, 1611.79, 1591.6, 1490.31, 1330.62, 1219.51, 1161.78, 1098.6,1069.5, 836.72, 797.57, 697.48.