Innate Immune Function of Mitochondrial Metabolism
10
0
0
Texte intégral
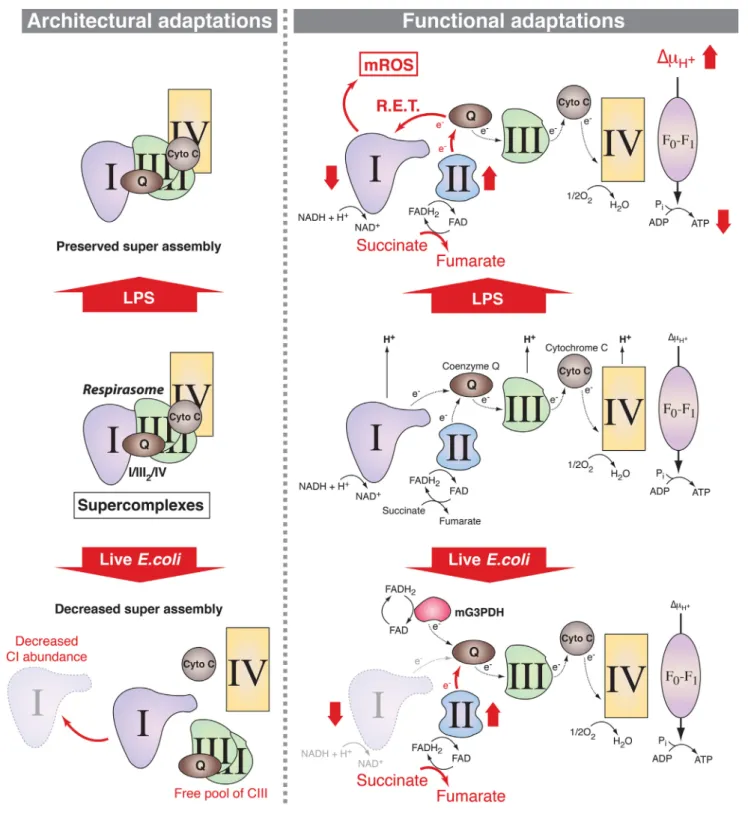
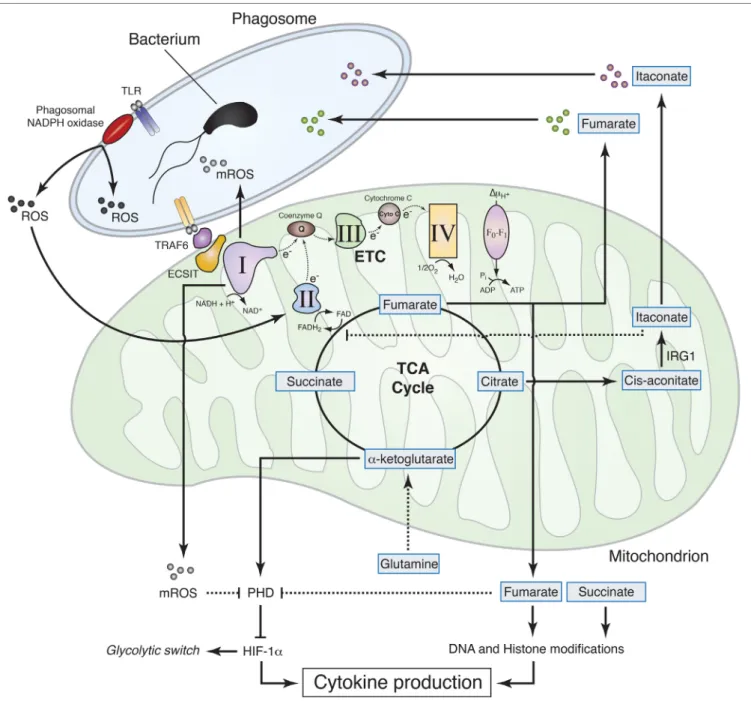
Figure
Documents relatifs
