Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome
11
0
0
Texte intégral
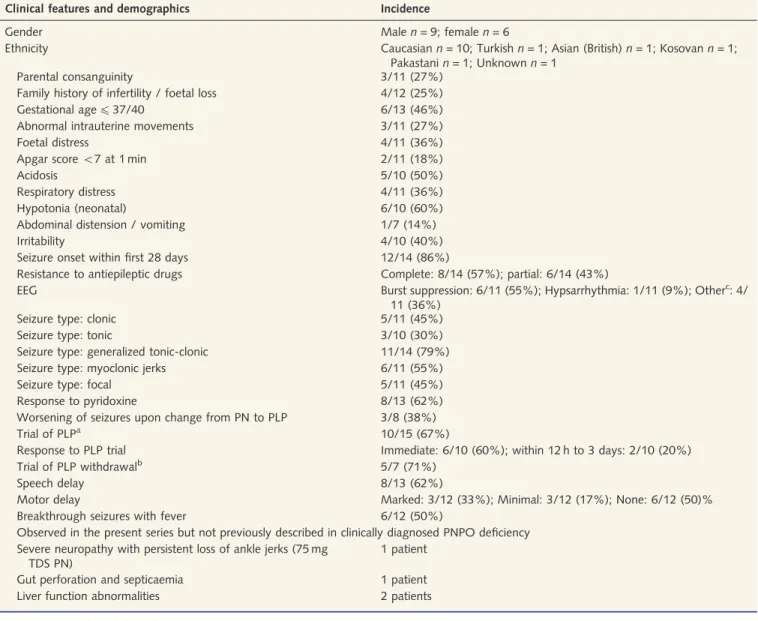
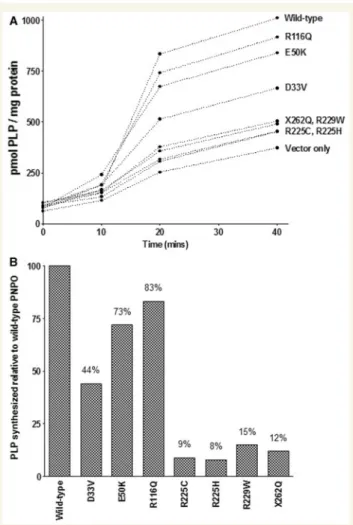
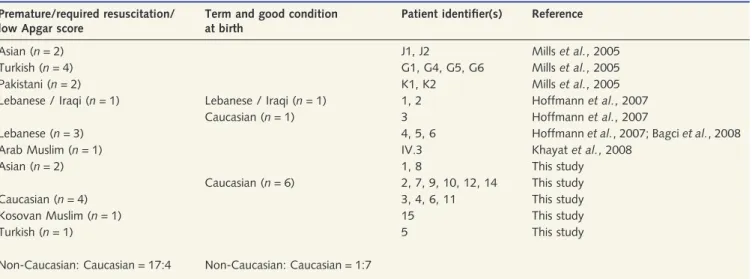
Figure
Documents relatifs