ROYAUME DU MAROC
UNIVERSITE MOHAMMED-V DE RABAT
FACULTE DE MEDECINE ET DE PHARMACIE DE RABAT
2020
Mémoire de Fin de Spécialité
-Pharmacie Industrielle-
Validation des procédés de fabrication
d’une spécialité pharmaceutique suite à un
changement d’équipement et augmentation
de taille de lot
Dr. Majda BENABBES
Remerciement
Ma gratitude s’adresse à Monsieur le Professeur Younes Rahali pour son
encadrement, son orientation, ses conseils et la disponibilité qu’il a témoigné.
TABLE DES MATIERES
Introduction ... 6
Partie I : Revue de littérature-Validation des procédés de fabrication ... 7
I. Définitions ... 7
II. Validation des procédés de fabrication : Réglementation, types et approches ... 7
1. Réglementation ... 7
1.1. International Conference of Harmonization (ICH) ... 7
1.2. Les Bonnes Pratiques de Fabrication Européennes ... 8
1.2.1. Chapitre 5 « Production » ... 8
1.2.2. Annexe 15 « Qualification et Validation » ... 8
1.3. Guideline de l’European medicine Agency ... 9
2. Les différents types de validation ...10
2.1. Validation prospective ...10
2.2. Validation concomitante (Simultanée) ...10
2.3. Validation rétrospective...10
3. Les différentes approches de validation des procédés ...10
3.1. L’approche traditionnelle de la validation ...10
3.2. L’approche par vérification en continu du procédé ...10
3.3. L’approche hybride ...11
III. Déroulement de la validation des procédés de fabrication ...12
1. Les prérequis à la validation des procédés ...12
2. Les documents support ...12
2.1. Plan Directeur de Validation ...12
2.2. Protocole de validation ...13
2.3. Rapport de validation ...13
3. La revalidation ...14
IV. Maitrise statistique des procédés ...14
1. Définition ...14
2. Les objectifs de la MSP ...14
3. La MSP et le processus de production ...15
4. Les cartes de contrôle ...15
4.1. Types de cartes de contrôle ...16
4.2. Etapes de la mise en œuvre des cartes ...16
5. Capabilité des procédés ...16
5.1. Dispersion des résultats ...18
5.3. Limites naturelles d’un processus ...19
5.4. Les indices de capabilité ...19
5.5. Décision sur la production ...20
Partie II : Application à la validation du procédé de fabrication d’une forme pâteuse « Crème » ...22
I. Projet de scale up et changement d’équipement de fabrication de la crème ...22
II. Validation du procédé des formes pharmaceutiques semi-solides ...22
1. Les paramètres critiques à valider en cours de production ...22
1.1. La température ...22
1.2. Taux de chauffage et de refroidissement ...23
1.3. Méthodes et vitesses de mélange ...23
1.4. Temps de mélange ...23
1.5. Débit (vitesse de pompage) ...24
1.6. Ajout de polymères et de gommes ...24
III. Matériels et méthodes ...24
1. Procédé de fabrication ...24
2. Validation du procédé de fabrication ...26
3. Validation du procédé de répartition (conditionnement primaire) ...28
4. Plan d’échantillonnage ...29
5. Validation du conditionnement primaire et secondaire ...30
IV. Résultats et discussion ...32
1. Description du procédé de fabrication ...32
2. Validation du procédé de fabrication ...33
3. Validation du procédé de répartition ...40
4. Validation du conditionnement primaire et secondaire ...48
Conclusion ...50
INDEX DES FIGURES
Figure 1 : Exemple d’une carte de contrôle Figure 2 : Dispersion à six écarts types
Figure 3 : Capabilité de deux procédés au regard de l’intervalle de tolérance et de la dispersion Figure 4 : Dispersion en cloche
Figure 5 : Schéma illustrant un processus « sous contrôle » et un processus « hors contrôle ». Figure 6 : Dispersion en cloche
Figure 7 : Représentations graphiques des différentes possibilités de combinaison entre les valeurs
des indices Cp et Cpk
Figure 8 : schéma du process de fabrication de la crème
Figure 9 : Schéma du Process de fabrication de la crème avec les paramètres du contrôle Figure 10 : Capabilité du procédé de fabrication-teneur en acide sorbique
Figure 11 : Capabilité du procédé de fabrication-teneur en parahydroxybenzoate de méthyle Figure 12 : Capabilité du procédé de fabrication-teneur en propyle gallate
Figure 13 : Capabilité du procédé de fabrication-teneur en substance active Figure 14 : Carte de contrôle de la masse moyenne des 3 lots de validation
INDEX DES TABLEAUX
Tableau 1 : Table de décision
Tableau 2 : liste des équipements utilisés pour la fabrication de la crème Tableau 3 : formule qualitative et quantitative des matières premières
Tableau 4 : formule qualitative et quantitative des articles de conditionnement Tableau 5 : IPC au cours de la préparation de la phase aqueuse
Tableau 6 : IPC au cours de la préparation de la phase grasse
Tableau 7 : IPC au cours de la préparation de la solution du principe actif Tableau 8 : IPC durant la validation du mélange final
Tableau 9 : IPC durant la validation du stockage
Tableau 10 : échantillonnage pour la validation du procédé de répartition (Par journée de
répartition)
Tableau 11 : échantillonnage pour la validation du procédé de répartition (par lot) Tableau 12 : Contrôle durant la validation du procédé de répartition
Tableau 13 : plan d’échantillonnage pour la validation du procédé de fabrication de la crème Tableau 14 : table de décision des défauts du conditionnement secondaire
Tableau 15 : Niveau de qualité acceptable des défauts du conditionnement secondaire Tableau 16 : table de décision des défauts du conditionnement secondaire
Tableau 17 : Niveau de qualité acceptable des défauts du conditionnement primaire Tableau 18 : Résultats du contrôle du pH
Tableau 19 : Résultats du contrôle de la teneur en eau
Tableau 20 : Résultats du contrôle de la teneur en acide sorbique
Tableau 21 : Résultats du contrôle de la teneur en parahydroxybenzoate de méthyle Tableau 22 : Résultats du contrôle de la teneur en gallate de propyle
Tableau 23 : Résultats du contrôle de la teneur en substance active Tableau 24 : critère d’acceptation pour le rendement du mélange final Tableau 25 : Résultats du contrôle du pH
Tableau 26 : Résultats du contrôle de la teneur en eau
Tableau 27 : Résultats du contrôle de la teneur en substance active Tableau 28 : Résultats du contrôle de la teneur en acide sorbique
Tableau 29 : Résultats du contrôle de la teneur en parahydroxybenzoate de méthyle Tableau 30 : Résultats du contrôle de la teneur en gallate de propyle
Tableau 31 : Résultats du contrôle des masses moyennes des 3 lots de validation Tableau 32 : Résultats du contrôle du pH
Tableau 34 : Résultats du contrôle de la teneur en substance active Tableau 35 : Résultats du contrôle de la teneur en acide sorbique
Tableau 36 : Résultats du contrôle de la teneur en parahydroxybenzoate de méthyle Tableau 37 : Résultats du contrôle de la teneur en matières extractibles
Tableau 38 : Résultats du contrôle de la teneur en gallate de propyle Tableau 39 : Résultats de la recherche de la propreté microbienne
Tableau 40 : Résultats du rendement du conditionnement secondaire et du produit fini Tableau 41 : Résultats du contrôle du conditionnement secondaire
Tableau 42 : Résultats du rendement du conditionnement primaire Tableau 43 : Cadence de la remplisseuse
6 | P a g e
Introduction
Dans un secteur où la rigueur et la précision sont demandées, il convient selon les bonnes pratiques de fabrication (BPFs) de mettre en place un système qualité pharmaceutique adéquat pour répondre aux exigences des autorités de santé et aux besoins des patients, c’est à ce moment que la validation intervient. Il s’agit d’un exercice réglementaire et l’un des principaux outils de l’assurance qualité en industrie pharmaceutique, qui est définit comme une démonstration assurant, avec un grand degré de certitude et preuves à l’appui, qu’un procédé permettra d’atteindre les résultats escomptés, de façon uniforme et continue.
Les études de validation sont réalisées pour les essais analytiques, le matériel, les systèmes de ventilation, d’adduction d’eau et de vapeur dans les établissements et pour des méthodes, comme les procédés de fabrication, le nettoyage, la stérilisation, le remplissage stérile ou la lyophilisation… Plus particulièrement, la validation des procédés de fabrication constitue une des étapes cruciales et indispensables à la production de médicaments de qualité, efficaces et surs. C’est un élément clé pour qu’un médicament soit commercialisé puisqu’elle reflète la capacité d’un fabricant à maitriser et contrôler toutes ses opérations de fabrication critiques ainsi que leurs sources de variabilité. C’est aussi une démarche de progrès qui, par une meilleure connaissance et une meilleure maitrise des procédés, permet une diminution des coûts de production et de contrôle.
En quelques années, la validation de nettoyage est ainsi devenue un sujet d’actualité dans l'industrie pharmaceutique soumise aux Bonnes Pratiques de Fabrication (BPF). Le nettoyage des locaux et des équipements doit donc être impliqué dans la démarche qualité de l'entreprise au même titre que les autres phases de fabrication et, ainsi, être maîtrisé.
Ce mémoire aura donc comme finalité de présenter un travail sur la validation du procédé de fabrication qui s’inscrit dans le cadre d’un projet de changement d’équipement de production des formes pâteuses (crèmes et gels) et d’augmentation de taille de lot.
Dans un premier temps, nous nous intéresserons plus précisément à la réglementation et des recommandations associées à la validation des procédés de fabrication puis à la démarche globale de mise en place. Dans un deuxième temps nous détaillerons un cas pratique dans le cadre de la production des formes pâteuses sur le site de production de Maphar à Casablanca-Zenata.
7 | P a g e
Partie I : Revue de littérature-Validation des procédés de fabrication
I.
Définitions
Procédé de fabrication : un procédé de fabrication correspond à une succession d’étapes
déterminées qui vont permettre d’obtenir un produit (Output) à partir de différents éléments entrants (Inputs) en suivant différents paramètres. Dans le domaine pharmaceutique :
Les inputs correspondent aux : Les matières premières,
Les équipements de production, Le personnel.
Les outputs correspondent aux produits finis.
Validation : L’établissement de la preuve, en conformité avec les principes de bonnes pratiques de
fabrication, que la mise en œuvre ou l’utilisation de tout processus, procédure, matériel, matière première, article de conditionnement ou produit, activité ou système permet réellement d’atteindre les résultats escomptés. Le terme de validation est notamment utilisé pour les procédés de fabrication, de nettoyage, les systèmes informatisés et les méthodes analytiques [1].
II.
Validation des procédés de fabrication : Réglementation, types et
approches
La validation des procédés est un élément essentiel des BPF. Elle constitue un aspect clé du système qualité et de l’évaluation des dossiers pour l’autorisation de mise sur le marché. L’ensemble des réglementations suivantes vont fournir, aux industriels, les outils nécessaires pour construire une approche de validation assurant la composition, la qualité, l’efficacité et la pureté des produits fabriqués, en prévenant les contaminations, les mix-up, mes déviations, les erreurs et défaillances.
1. Réglementation
1.1. International Conference of Harmonization (ICH)
L’ensemble de ces guides de Q7 à Q11 décrivent une nouvelle approche de la qualité fondée sur le cycle de vie du produit et la gestion des risques.
L’ICH Q7 correspond aux BPF dans la production des principes actifs chimiques et biologiques. En effet, les exigences concernant la fabrication des principes actifs et des produits finis ne sont pas les mêmes. Ces deux types de fabrication font chacune l’objet d’une section dans le dossier réglementaire du médicament.
L’ICH Q8 et Q11 encadrent respectivement les activités de développement du produit fini et de développement du principe actif. L’ICH Q11 étant la dernière directive publiée, celle-ci intègre les aspects développés dans les ICH Q7 à Q11.
8 | P a g e
L’ICH Q9 définit les principes de la gestion des risques et l’ICH Q10 décrit un modèle de système qualité pharmaceutique qui peut être appliqué à l’ensemble du cycle de vie d’un produit. Il va donc au-delà des exigences actuelles des BPF qui ne s’appliquent pas au développement d’un produit. Les spécifications qui vont au-delà des exigences BPF restent optionnelles [2-6].
1.2. Les Bonnes Pratiques de Fabrication Européennes
Selon les BPF Européennes, la validation du procédé est définie comme étant « la preuve documentée que le procédé mis en œuvre à l'intérieur des paramètres établis peut fonctionner de manière efficace et reproductible pour fabriquer un produit conforme à ses spécifications et à ses caractéristiques de qualité préétablies » [1].
1.2.1. Chapitre 5 « Production »
Dans ce chapitre, 4 recommandations font référence à la validation :
- « Les études de validation doivent conforter les bonnes pratiques de fabrication ; elles doivent être menées conformément à des procédures définies. Les résultats et les conclusions doivent être consignés ».
- « Lors de l'adoption d'une nouvelle formule de fabrication ou d'une nouvelle méthode de préparation, il convient de démontrer qu'elle satisfait à la production de routine et que le processus choisi, avec les produits et le matériel prévus, donne systématiquement un produit de la qualité requise ».
- « Il convient de valider toute modification importante du processus de fabrication, y compris au niveau du matériel ou des produits, lorsque cette modification peut affecter la qualité du produit ou la reproductibilité du processus ».
- « Les procédés et les procédures doivent être périodiquement soumis à une nouvelle validation critique en vue de confirmer leur aptitude à conduire aux résultats escomptés ».
Ces recommandations encadrent donc la manière dont doit être réalisée la validation d’un procédé de fabrication. Les approches à suivre sont plus détaillées dans l’annexe 15 « Qualification et validation » [1].
1.2.2. Annexe 15 « Qualification et Validation »
La présente annexe mise à jour en Janvier 2017 par l’ANSM, décrit les principes de la qualification et de la validation applicables à la fabrication des médicaments : « Les BPF stipulent que le fabricant doit contrôler les aspects critiques des opérations qu’il met en œuvre au moyen de qualification et de validation tout au long du cycle de vie du produit et du procédé. Tout changement planifié relatif aux installations, aux équipements, aux utilités et aux procédés, susceptible d’avoir un impact sur la qualité du produit, doit être formellement documenté, et l’impact sur le statut de validation ou la
9 | P a g e
stratégie de contrôle évalué ». Elle met en avant l’importance de réaliser un suivi en continu du procédé et du produit tout au long de leur cycle de vie puisque ceux-ci sont intimement liés.
Cette annexe décrit les documents relatifs à l’activité de validation et donne des définitions sur les différentes approches de la validation (prospective, concomitante, traditionnelle, vérification en continue du procédé, hybride) [1].
1.3. Guideline de l’European medicine Agency
L’EMA revient sur les différentes approches utilisées pour valider le procédé de fabrication de produits finis. Elle peut également s’appliquer aux substances actives.
Elle décrit l’approche de vérification continue du procédé qui est définie dans l’ICH Q8 comme étant « une approche alternative à la validation du procédé traditionnelle dans laquelle la performance du procédé est continuellement surveillée et évaluée ».
A l’instar de la validation traditionnelle qui vise à démontrer que le procédé fournit un produit de la qualité requise sur 3 lots successifs à l’échelle commerciale et une fois que la phase de développement est achevée, la vérification continue est initiée pendant le développement (en général avant la fabrication des lots cliniques de phase III) et se poursuit pendant tout le cycle de développement et de vie du produit. Dès lors, la validation n’est plus basée sur 3 lots mais sur tous les lots qui ont été produits depuis l’initiation de la vérification continue, étape à laquelle le fabricant considère que son procédé est sous contrôle. Cette vérification continue du procédé peut être réalisée à l’aide de technologies innovantes telles que les Technologies d’Analyse du Procédé (PAT) et d’outils statistiques permettant de suivre dans le temps la capacité du procédé à respecter des spécifications (Statistical process control).
De ce fait, toutes les données générées pendant la vérification continue sont intégrées au dossier de soumission. Elles sont donc issues à minima des études réalisées à l’échelle du laboratoire ou de l’échelle pilote. Cette approche ne peut, cependant, être envisagée que si le médicament a été développé selon l’approche Quality By Design ou si les connaissances acquises pendant le développement permettent de justifier son utilisation. Elle peut être utilisée pour valider soit l’ensemble du procédé soit les étapes de production que le fabricant considère comme critiques, ce dernier cas correspond à l’approche hybride puisque les étapes considérées comme non critiques seront validées selon une approche traditionnelle[7].
10 | P a g e
2. Les différents types de validation 2.1. Validation prospective
C’est une validation réalisée avant la production de routine de produits destinés à la vente. Elle concerne soit un nouveau produit ou un produit fabriqué selon un procédé de fabrication modifié, où les modifications sont importantes et peuvent se répercuter sur la qualité du produit[1].
2.2. Validation concomitante (Simultanée)
La validation a lieu pendant la fabrication de routine de produits destinés à la vente. Elle doit rester exceptionnelle et s’applique dans le cas de productions peu fréquentes notamment pour les produits à forte valeur ajoutée tels que les médicaments orphelins ou dans le cas de modifications de procédé mineures. Cette décision doit être justifiée, documentée et approuvée [1].
2.3. Validation rétrospective
Il s’agit d’une validation réalisée pour un produit déjà commercialisé et basée sur les données historiques de production. Actuellement, cette approche n’est plus autorisée [1].
3. Les différentes approches de validation des procédés 3.1. L’approche traditionnelle de la validation
Dans l'approche traditionnelle, le développement pharmaceutique est essentiellement empirique et les études menées lors du développement sont souvent réalisées avec une seule variable à la fois. Dans l’approche traditionnelle, la validation du procédé est habituellement basée sur la production de trois lots successifs à l’échelle commerciale. Elle doit notamment permettre de s’assurer de l'uniformité et de l'intégrité du lot et faire appel aux deux types de validations décrites précédemment : La validation prospective et la validation concomitante.
Ces essais doivent démontrer que le procédé mis en œuvre à l’intérieur des paramètres préétablis doit fournir de manière reproductible un produit de la qualité requise.
Dans l'approche traditionnelle, les contrôles comme, par exemple, les contrôles en cours de fabrication sont fixes et ne sont pas censés être modifiés au cours du temps. De plus, la stratégie de contrôle repose sur un échantillonnage très important et principalement sur l'analyse des produits intermédiaires et finis [1, 7].
3.2. L’approche par vérification en continu du procédé
La vérification en continu des procédés ou Continuous Process Verification est reconnue comme nouvelle approche de validation des procédés de fabrication intégrant le concept de lifecyle. La validation est désormais ainsi réalisable depuis le développement jusqu’à la production commerciale.
11 | P a g e
Cette approche est applicable pour les produits développés selon une approche de qualité par la conception QbD (Quality by design), où il a été scientifiquement établi pendant le développement que la stratégie de contrôle génère un niveau élevé d’assurance de la qualité du produit, une vérification en continu du procédé peut alors être utilisée comme une alternance à la validation classique.
Selon la ligne directrice ICH Q8, le Quality By Design (QbD) ou qualité par la conception est définie comme « une approche du développement qui commence par des objectifs prédéfinis et met l'accent sur la compréhension des produits et des procédés, et leurs contrôles, fondés sur des preuves scientifiques et la gestion du risque qualité ». Cela implique de prendre et d'utiliser les connaissances acquises lors du développement des procédés, de comprendre les sources de variation multiples et leur impact sur le procédé et par conséquent, sur la qualité du produit (par exemple, la glycosylation, les formations d'agrégats ou la présence d’impuretés, etc.). La stratégie de contrôle qui en découlera sera fondée sur des connaissances scientifiques solides et permettra de garantir que le procédé fournit de façon reproductible un produit de qualité.
Avec cette approche, le fabricant montre que les différentes variables qui peuvent influencer le procédé et la qualité du produit sont sous contrôle. L’ICH Q8 est également applicable, en plus des produits finis, au développement et à la fabrication de substance active[3].
Toutes les données produites au cours des études réalisées à l’échelle laboratoire, l'échelle pilote et/ou l'échelle commerciale, seront utilisées pour assurer la production d'une substance active et d'un médicament de qualité requise. Ce sont ces données qui démontreront donc si l'échelle commerciale est capable de fournir un produit d’une qualité acceptable
L'objectif principal de la QbD est donc de développer un procédé robuste et bien compris. Ceci peut être réalisé grâce à l’utilisation de nouvelles technologies telles que les outils de Design of Experiments, les technologies d’analyse du procédé (Process Analytical Technologies ou PAT), les outils statistiques qui permettent de contrôler le procédé et à la définition d’un espace de conception (Design Space) [7-9].
3.3. L’approche hybride
Une approche hybride mêlant l’approche traditionnelle et la vérification en continu du procédé peut être utilisée quand les connaissances du produit et la compréhension du procédé issues de l’expérience de fabrication et des données historiques des lots sont suffisantes. Cette approche peut aussi être utilisée pour des activités de validation après modification ou pendant le suivi en continu du procédé, même si le produit a été validé à l’origine avec une approche traditionnelle [7].
12 | P a g e
III. Déroulement de la validation des procédés de fabrication
1. Les prérequis à la validation des procédés
Avant de pouvoir valider un procédé de fabrication, différents éléments sont requis :
- La validation doit être conduite conformément à un protocole détaillé, préétabli ou à une série de protocoles, qui est à son tour soumis à des procédures de maitrise des changements ;
- le personnel conduisant les études ainsi que les opérateurs doivent être convenablement formés, motivés, qualifiés et compétents pour exécuter leur tâche ;
- Toutes les données générées pendant les études antérieures doivent être revues et approuvées et évaluées contre des critères prédéterminés ;
- Des installations appropriées, un équipement, des instruments doivent être qualifiés et disponibles ; - Une documentation complète devrait être disponible pour définir un support et enregistrer la validation complète du procédé [1].
2. Les documents support
Un programme de validation efficace doit s’accompagner d’une documentation étendue du début du développement du médicament à l’échelle de production. La documentation progressera jusqu’à la production à grande échelle, fournissant une histoire du produit aussi complète que possible. Elle sera constituée :
Plan directeur de validation (Validation Master Plan), Procédure de validation,
Protocole de validation, Rapport de validation,
Ainsi que d’autres documents critiques se rapportant à la formulation, au procédé et au développement de méthodes analytiques. L’ensemble contiendra une histoire complète du produit final fabriqué.
2.1. Plan Directeur de Validation
Le plan directeur de validation est le document qui permet de décrire la stratégie et l’approche de validation qui sera suivie pour un projet donné. Il reprend en général les différents points suivants :
La politique de validation,
La structure organisationnelle des activités de validation,
Le relevé des installations, systèmes, équipements et procédés à valider, Le format de la documentation,
La planification et programmation, La maîtrise des changements,
13 | P a g e
2.2. Protocole de validation
La rédaction d’un protocole de validation est une exigence réglementaire qui va spécifier la façon
dont la validation d’un procédé particulier doit être conduite. Il décrit l’ensemble des opérations à réaliser, les tests à effectuer et les critères d’acceptation. Un protocole de validation doit contenir au minimum les éléments suivants :
L’objectif et le contenu de la validation,
Les responsabilités concernant l’exécution, la réalisation des analyses, l’approbation, etc… La description du procédé de fabrication,
L’identification des lots et la justification du nombre de lots réalisés, L’identification des équipements et des installations utilisées,
L’identification des matières premières utilisées,
Les paramètres critiques du procédé et les critères d’acceptation, Les IPC à réaliser et leurs spécifications,
Le plan de prélèvement,
Les méthodes d’essais analytiques utilisées, Les études de stabilité devant être réalisées,
La conclusion statuant sur le déroulement de la validation [12].
2.3. Rapport de validation
Toutes les données recueillies durant la phase de validation doivent être compilées au sein d’un rapport de validation. Ce document permet de statuer sur la validation du procédé. Y sont consignés les résultats et commentaires obtenus concernant la fabrication proprement dite, les In-Process Controls (IPC), les tests réalisés sur le produit fini. Pour une meilleure compréhension, les différentes données pourront être présentées sous formes de graphiques ou encore de tableaux. Ces résultats sont ensuite évalués et analysés en les comparants aux limites et critères d’acceptation définis dans le protocole.
Le rapport doit également comporter toutes les modifications et les déviations rencontrées durant la réalisation des lots de validation, incluant leurs investigations, leur conclusion et les actions correctives et préventives prises. Les premières données concernant les études de stabilité peuvent être intégrées au rapport. Les dossiers de lots pourront également être joints à ce rapport. Enfin, une conclusion finale vis-à-vis du statut de la validation sera rédigée. Elle prendra en compte tous les résultats et constatations précédemment évalués [11].
14 | P a g e
3. La revalidation
La revalidation fournit l'évidence que les changements dans un procédé et / ou son environnement, introduits intentionnellement ou involontairement, n'affectent pas négativement le procédé et la qualité du produit. Il existe deux catégories principales de revalidation :
La revalidation lorsqu’il y a eu un changement au niveau du procédé (ex : dans le cas d’un transfert de procédé d’un laboratoire pharmaceutique à un autre ou d’un site à un autre site). La revalidation périodique à intervalles réguliers. En effet, même des changements mineurs
qui individuellement ne requièrent pas de revalidation peuvent, par effet cumulatif sur une période déterminée, affectés l’état validé du procédé.
Des procédures devraient donc être mises en place afin de traiter ces deux cas.
Les changements qui peuvent nécessiter d’entreprendre de nouvelles activités de validation sont les suivants :
Changement qualitatif ou quantitatif de la formule,
Changement des matières premières et des articles de conditionnement, Changement au niveau des spécifications du produit,
Changement des équipements de production, des locaux, des systèmes et des utilités, Changement des paramètres opératoire du procédé [13, 14].
IV.
Maitrise statistique des procédés
1. Définition
La MSP est un élément d’assurance qualité, c’est un ensemble d’actions qui vise à évaluer et maintenir un processus de production en état de fabriquer des produits conformes aux spécifications et avec des paramètres stables dans le temps. Son objectif est de maitriser un processus mesurable par suivi graphique temporel basé sur des fondements statistiques.
Il s’agit d’une méthode de surveillance d’un processus afin d’identifier des causes spécifiques de variation et signaler le besoin de prendre des actions correctives, quand c’est approprié [15, 16].
2. Les objectifs de la MSP
La MPS peut être utilisé à différentes étapes du procédé pour analyser ses variations avec comme objectifs réduire et maitriser les variations.
La méthode MPS est devenue un outil de compétitivité sans équivalent et qui vise à: Garantir une même qualité du produit,
Assurer la stabilité dans le temps,
15 | P a g e
3. La MSP et le processus de production
Ainsi défini précédemment, un processus désigne l’ensemble des moyens et activités liées qui transforment des éléments entrants en éléments sortants. Le contrôle en cours de production a pour but d’obtenir une production stable avec un minimum de produits non conformes aux spécifications. Le contrôle de la qualité est ‘dynamique’ : il ne s’intéresse pas au résultat isolé et instantané, mais au suivi dans le temps : il ne suffit pas qu’un paramètre soit dans les limites des spécifications, il faut aussi surveiller la répartition chronologique des pièces à l’intérieur des intervalles de tolérances.
Lamaitrisestatistiquedesprocédésvisantàuneproductioncentréeetlamoinsdisperséepossible.
Processus de production=Ensemble du processus de fabrication+ Processus de contrôle. Les procédés sont incapables de produire toujours exactement le même produit. Lorsqu’on contrôle une des caractéristiques d’un produit, on observe une dispersion des valeurs mesurés autour de la valeur cible. Il existe une variation dite naturelle au procédé qui fait que la qualité varie : c’est la variation normale. Cette variation se répercute sur la qualité du produit mais dans des proportions acceptables. Elle reste à l’intérieur des limites naturelles du procédé. Elle est inhérente au procédé et est souvent difficile à réduire sans toucher au procédé lui-même
A côté de cette variation naturelle, il existe un autre type de variation lié à des causes spéciales qui vient s'ajouter à la variation naturelle : c'est la variation anormale. Cette dernière pousse les paramètres du procédé à sortir des limites de contrôle. Pour revenir à l'intérieur des limites, le procédé attend que les causes spéciales soient analysées pour être corrigés.
Les causes probables pour cette variation anormale sont : Machine, Main d'œuvre, Matériau, Milieu et Méthode (les 5 M) [15].
4. Les cartes de contrôle
Une carte de contrôle est un outil permettant de déterminer le moment où apparait une cause assignable entrainant une dérive du processus de fabrication. Ainsi, le processus sera arrêté au bon moment, c’est-à-dire avant qu'il ne produise des produits non conformes (hors de l'intervalle de tolérance).
Une carte de contrôle est un graphique reflétant le déroulement du procédé de fabrication. Suite aux prélèvements effectués en cours de production, les résultats des analyses effectuées sur ces échantillons vont être reportés sur le graphique. Les cartes de contrôle permettent la surveillance de la fabrication, en s’assurant que les attributs et les paramètres contrôlés restent stables ou conformes aux spécifications, en tenant compte d’une certaine variabilité inévitable.
Cet outil se présente comme un graphique dont les points représentent le suivi dans le temps d'une caractéristique du processus dont la valeur centrale (souvent la moyenne) est représentée par une
16 | P a g e
ligne horizontale ainsi que la limite de contrôle inférieure (LIC), et la limite de contrôle supérieure (LIS).
Ces deux valeurs sont les limites à l'intérieur desquelles le processus est sous contrôle. Les valeurs de la caractéristique contrôlée doivent se trouver à l'intérieur de ces limites, sinon ces valeurs sont 'hors contrôle' et doivent être examinées (Figure1) [15-18].
Figure 1 : Exemple d’une carte de contrôle 4.1. Types de cartes de contrôle
Les cartes de contrôle aux mesures : elles s’appliquent à des valeurs continues telles que le poids, le volume, le dosage…, etc. Elles donnent une valeur mesurée plus riche d’information que le simple constat bon ou mauvais.
Les cartes de contrôle aux attributs : Les attributs sont des données fondées sur deux valeurs seulement (conforme/non conforme). La technique des cartes de contrôle aux attributs, avec le même type de calcul des limites que les cartes aux mesures, sont intéressante car elle permet de suivre les progrès réalisés en cours de production.
Par contre les cartes aux attributs ne donnent pas d’avertissement, en cas de changement dans le procédé, avant la production d’un nombre accru de non conformes [17, 18].
4.2. Etapes de la mise en œuvre des cartes
- Choix des caractéristiques à suivre, - Choix du type de contrôle,
- Choix du type de carte (en fonction de la rapidité du déréglage),
- Choix de l'échantillonnage (détermination de l'effectif et de la fréquence d'échantillonnage), - Etude préliminaire du processus (détermination des paramètres de la caractéristique suivie), - Etablissement des règles de décision [15].
5. Capabilité des procédés
17 | P a g e
compatibles avec la qualité attendue. Pour un procédé, l’étude de capabilité compare l’intervalle de tolérance à la dispersion, c’est-à-dire la performance demandée à la performance réellement obtenue. Lors de l’étude d’un procédé, le terme capabilité désigne son aptitude.
L’intervalle de tolérance est défini par la distance comprise entre les tolérances supérieure et inférieure. Si les valeurs obtenues à partir des échantillons dépassent ces tolérances, les échantillons sont considérés comme non-conformes.
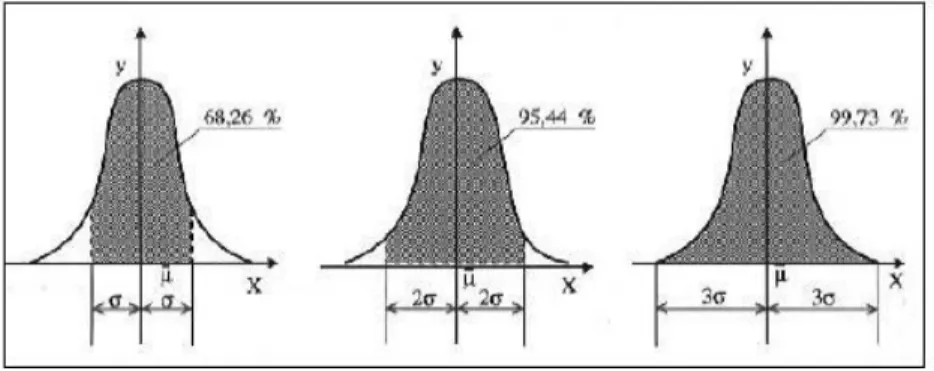
Pour le paramètre étudié, la dispersion du procédé est définie par le calcul de son écart type. La dispersion à « six écarts types » (6 σ) correspond à l’intervalle contenant 99,73% des valeurs du paramètre étudié. Ceci dit, la probabilité de trouver une valeur comprise entre+/-trois écarts types est de 0,9973, soit 99,73% [19, 20].
Figure 2 : Dispersion à six écarts types
Plus la dispersion du paramètre sera faible, plus l’écart type sera réduit et plus le risque de dépasser les tolérances sera faible ou si l’on préfère, plus l’écart type sera réduit, plus la production sera conforme.
Figure 3 : Capabilité de deux procédés au regard de l’intervalle de tolérance et de la dispersion
Le premier procédé présente une dispersion supérieure aux tolérances. Il en résulte un pourcentage certain de production non-conforme.
18 | P a g e
l’intervalle de tolérance. Toute la production est conforme. 5.1. Dispersion des résultats
L'analyse des productions sur une machine montre que, en l'absence de déréglage, la répartition des produits suit une courbe en cloche selon une loi : la loi normale.
Désormais, lorsque nous parlerons de la production d'une machine, nous la modéliserons par une courbe en cloche, dont les deux caractéristiques importantes seront la position et l'échelle.
La position moyenne (notée 𝑋̅) des pièces donne une bonne indication de la position de réglage de la machine. µ représente la moyenne de l'échantillon.
Pour mesurer l’importance des variations autour de la moyenne, il suffit de mesurer la largeur de base de la courbe. La largeur de base de la courbe est appelée : dispersion. Cette base de la courbe est notée 6 σ, dont σ est l’écart type de la population (Figure 4).
Figure 4 : Dispersion en cloche
Il y a quelques exceptions comme les défauts de forme ou les défauts de position où il est normal de ne pas obtenir une courbe en cloche, dans ce cas on peut distinguer deux causes :
- Causes communes : L'ensemble de ces causes communes forme la variabilité intrinsèque du processus.
- Causes spéciales : Ce sont les causes de dispersion identifiables, souvent irrégulières et instables, et par conséquent difficiles à prévoir. L’apparition d’une cause spéciale nécessite une intervention sur le processus. Contrairement aux causes communes, les causes spéciales sont en général peu nombreuses.
5.2. Processus « sous contrôle » et « hors contrôle »
Un processus « sous contrôle » est un processus dans lequel seules subsistent les causes communes. La répartition de la production suit alors une courbe en cloche et elle est centrée sur la cible.
Un processus « hors contrôle » est soumis à la présence de causes spéciales. Le résultat de la production ne suit donc pas nécessairement une courbe en cloche et la production peut être décentrée par rapport à la cible (Figure 5).
19 | P a g e
Figure 5 : Schéma illustrant un processus « sous contrôle » et un processus « hors contrôle ». 5.3. Limites naturelles d’un processus
Si la moyenne de la production est centrée sur la cible, il est donc naturel de trouver des valeurs comprises entre ± 3 écarts types (σ) de cette cible. Les valeurs « cible + 3 σ » et « cible – 3 σ » représentent les limites naturelles du processus. Tant qu'une valeur est dans ces limites, il n'y a pas de raison d'agir sur le processus, on risquerait de décentrer un processus bien centré. Si une valeur sort de ces limites, on a une forte probabilité que le processus ne soit plus centré sur la cible, il faut alors le recentrer.
Figure 6 : Dispersion en cloche 5.4. Les indices de capabilité
La capabilité potentielle(Cp) : compare l’intervalle de tolérance (IT)des spécifications à la dispersion du procédé. Elle permet d’évaluer la variation du procédé.
𝑇𝑠 − 𝑇𝑖 𝐶𝑝= 6σ
Avec Ts : valeur supérieure de tolérance ; Ti : valeur inférieure de tolérance ; σ : écart-type.
La capabilité réelle (Cpk) : permet de mesurer le centrage du procédé par rapport à la valeur cible attendue. Pour caractériser la performance d’un procédé, le calcul de la capabilité potentielle n’est pas suffisant. En effet, l’étude de la capabilité potentielle ne reflète pas la tendance centrale du procédé, car deux procédés ayant une dispersion identique, peuvent se situer différemment au sein de l’intervalle de tolérance.
20 | P a g e
Avec X : moyenne
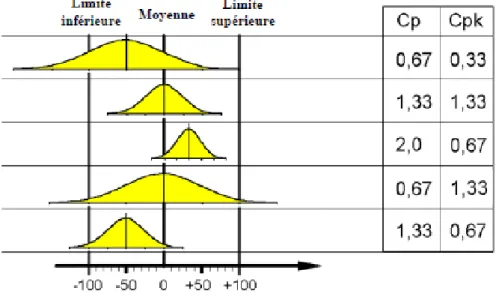
Il est important de prendre en compte les deux indices conjointement car ils sont complémentaires et permettent une compréhension et une analyse plus justes du procédé. Egénéral, les valeurs du Cp et Cpk doivent être supérieures ou égales à 1,33 pour que la dispersion et le centrage sur la moyenne des valeurs soient acceptables [15, 21].
Figure 7 : Représentations graphiques des différentes possibilités de combinaison
entre les valeurs des indices Cp et Cpk
5.5. Décision sur la production
En fonction de l'échantillonnage qui a été réalisé, il faut décider si la production peut être acceptée ou si elle doit donner lieu à un tri. Pour prendre cette décision, il faut tenir compte de la capabilité court terme du processus exprimé par le Cp. Le tableau 1 donne les règles à appliquer. En cas de mauvaise capabilité (Cp < 1,33), on doit systématiquement trier la production pour obtenir un résultat correct. En cas de bonne capabilité (Cp > 1,67), on peut produire sans trier même si on constate un point hors contrôle. Dans les cas intermédiaires
21 | P a g e
(1,33 < Cp < 1,67), on ne trie la production que dans les cas où on constate un point hors contrôle [15, 21].
22 | P a g e
Partie II : Application à la validation du procédé de fabrication
d’une forme pâteuse « Crème »
I.
Projet de scale up et changement d’équipement de fabrication de la
crème
Le présent travail s’inscrit dans le cadre d’un projet de changement d’équipement de fabrication et d’augmentation de la capacité de production (scale up) d’un produit pharmaceutique. La mise en place d’un tel projet nécessite de calculer la durée nécessaire à sa réalisation et d’assurer un plan de production permettant de maintenir un stock suffisant pour les besoins du marché. L’augmentation des parts de marché dans l’industrie pharmaceutique entraine une croissance de la demande, ainsi le fabricant a tout intérêt à s’adapter et à être réactif. Une telle démarche passe notamment par une augmentation de volume des équipements impliqués dans la fabrication. La crème objet du changement se fabriquait en une taille de lot de 300 Kg qui passe à 429 Kg (plus de 10% d la taille initiale) avec un changement de l’équipement de production avec une nouvelle capacité de 1000 L au lieu de 700 L pour l’ancien équipement. Ces changements font appel à une validation du procédé de fabrication.
Cependant, l’augmentation de la taille de lot demande une planification et une mise en œuvre rigoureuse. D’un autre côté, les facteurs de changement d’échelle peuvent être complexes et difficiles à prévoir. En effet, plus le procédé de fabrication est complexe, plus le changement d’échelle sera complexe.
II.
Validation du procédé des formes pharmaceutiques semi-solides
Les formes semi-solides constituent une proportion importante des formes pharmaceutiques. Les crèmes sont des formes posologiques semi-solides contenant une ou plusieurs substances médicamenteuses dissoutes ou dispersés dans une base appropriée. La consistance des semi-solides se situe entre le solide et le liquide ce qui constitue un défi pour les fabricants.
1. Les paramètres critiques à valider en cours de production 1.1. La température
Maintenir une température adéquate est essentiel au succès de la fabrication. Un échauffement excessif peut entraîner une dégradation chimique. Une température insuffisante peut entraîner des ruptures de charge et un refroidissement excessif pouvant entraîner la précipitation des ingrédients solubilisés.
23 | P a g e
L'étape d'émulsification d'une émulsion classique huile dans eau est un exemple de la nécessité d'un bon contrôle de la température. Si la température de la phase aqueuse est beaucoup plus basse que celle de la phase huileuse, les constituants fondus de la phase huileuse peuvent se solidifier lors de l'introduction dans la phase aqueuse ce qui empêche la formation de l’émulsion [22].
1.2. Taux de chauffage et de refroidissement
Un chauffage trop lent peut entraîner des rendements médiocres dus à la perte par évaporation. Un chauffage trop rapide peut brûler des zones du lot en contact avec la surface chauffante, ce qui augmente le potentiel de la matière brûlée dans le lot. Un refroidissement rapide peut entraîner une précipitation / cristallisation ou une augmentation de la viscosité [23].
1.3. Méthodes et vitesses de mélange
Il est essentiel de déterminer l’homogénéisation requise, les méthodes et les vitesses de mélange optimales. L'émulsification nécessite généralement une forte homogénéisation pour obtenir la taille et la dispersion optimales des gouttelettes, tandis que le mélange d'un gel peut nécessiter une faible homogénéisation afin de préserver certaines caractéristiques physiques, tel que la viscosité. Des vitesses de mélange appropriées doivent être obtenues pour chaque phase et à chaque étape de fabrication du lot.
Si le procédé implique uniquement un mélange à très faible homogénéisation, un polymère ne peut jamais être complètement dispersé, ce qui peut entraîner une viscosité non conforme aux spécifications [24].
1.4. Temps de mélange
Optimiser le temps de mélange nécessite la détermination du temps minimum requis pour la dissolution des composants et le temps de mélange maximum à ne pas dépasser afin d’éviter la défaillance du produit (par exemple, lorsque la viscosité commence à baisser).
Optimiser le temps de mélange nécessite d'identifier le temps minimum requis pour la dissolution des ingrédients et le temps de mélange maximum avant la défaillance du produit (par exemple, lorsque la viscosité commence à baisser). Pour les gels polymères, en particulier les types d’acide acrylique, le mélange excessif, particulièrement élevé, peut briser la structure du polymère. Dans une émulsion, le mélange excessif peut entraîner une séparation prématurée du produit, ce qui entraîne une diminution drastique de la viscosité.
24 | P a g e
1.5. Débit (vitesse de pompage)
Une émulsion eau dans huile peut nécessiter une vitesse d'addition plus lente qu'une émulsion huile dans eau classique, et le débit doit être modifié de manière appropriée. Des précautions doivent être prises pour tout procédé utilisant une pompe. Une sur-audition peut survenir si le produit est pompé trop rapidement. Si le pompage est trop lent, le produit subira un temps supplémentaire, exposant ainsi la formulation à une homogénéisation supplémentaire [25].
1.6. Ajout de polymères et de gommes
L'ajout de polymères (carbomères) et de gommes (xanthane) doit être effectué de manière très contrôlée si l'ajout est effectué directement dans le mélange. Il existe également d’autres méthodes d’incorporation : des éducteurs tels que les Tri-Blenders et les dispersants Quadro Ytron et la préparation d’une suspension de polymères ou de gomme dans un milieu peu ou pas soluble [26].
III. Matériels et méthodes
La validation du procédé de fabrication a été entreprise sur 3 lots ayant été fabriqués consécutivement dans les mêmes conditions. Il s’agit d’une validation prospective selon une approche traditionnelle.
1. Procédé de fabrication
Taille de lot : 429 Kg soit 28600 tubes de 15g.
a. Les équipements
Les équipements utilisés pour la fabrication de la crème sont qualifiés à la date de fabrication.
Tableau 2 : liste des équipements utilisés pour la fabrication de la crème Equipements
Cuve de fabrication (Mélangeur) Cuve double paroi 350 L Cuve de stockage Remplisseuse Encartonneuse
b. Composants
Les matières premières et les articles de conditionnements constituent l’input de la production, l’ensemble des composants sont contrôlés selon les BPF.
Les Matières premières
25 | P a g e
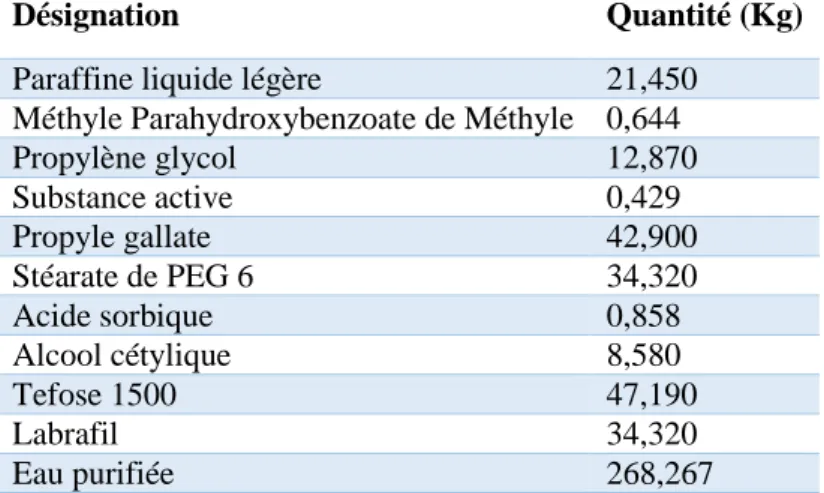
Désignation Quantité (Kg)
Paraffine liquide légère 21,450 Méthyle Parahydroxybenzoate de Méthyle 0,644
Propylène glycol 12,870 Substance active 0,429 Propyle gallate 42,900 Stéarate de PEG 6 34,320 Acide sorbique 0,858 Alcool cétylique 8,580 Tefose 1500 47,190 Labrafil 34,320 Eau purifiée 268,267
Les articles de conditionnement
Tableau 4 : formule qualitative et quantitative des articles de conditionnement Désignation Quantité (unité)
Tube Plastique 28886 Etui 28886 Notice 28886 Caisse américaine 360x260x146 286 Vignette autocollante 28886
c. Schéma du procédé de fabrication
Le procédé de fabrication passe par un ensemble d’étapes illustrées dans le schéma ci-dessous :
Opérations Etapes Matériel et paramètres
Fusion
1
Ceinture chauffante
Température = 200°C Température MP = 70°C
Préparation de la phase aqueuse 2
Cuve chauffante Double paroi 350L
Température= 73 à 77°C Vitesse d’agitation=……….
Durée d’agitation=30 min
Préparation de la phase grasse 3
Mélangeur (cuve de fabrication)
Vitesse d’homogénéisation = …… Vitesse d’agitation=……….
Durée d’agitation=15 min Température= 73 à 77°C
Emulsification 4
Mélangeur (cuve de fabrication)
Température phase aqueuse = 73 à 77 °C Température phase grasse = 73 à 77 °C
26 | P a g e
Vitesse d’agitation = ……. Durée =15 minutes
Vide =-0.5 bar
Préparation de la solution de principe de
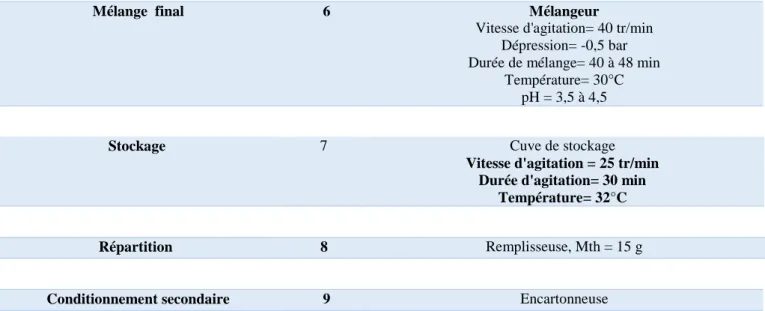
principe actif 5 Conge INOX 20 L Température de chauffage= 45 °C Durée =20 minutes Mélange final 6 Mélangeur Vitesse d'agitation= ………. Dépression= -0,5 bar Durée de mélange= 40 min
Température= 30°C pH = 3,5 à 4,5
Stockage 7
Cuve de stockage
Vitesse d'agitation = ………. Durée d'agitation= 30 min
Température= 30°C
Répartition (conditionnement primaire) 8 Remplisseuse, Mth = 15 g
Conditionnement secondaire 9 Encartonneuse
Figure 8 : schéma du process de fabrication de la crème 2. Validation du procédé de fabrication
2.1.Préparation de la phase aqueuse
Objectif : Vérifier la dissolution des matières premières.
Echantillonnage : Dans la cuve chauffante, après le temps d’agitation de 30 minutes, prélever un échantillon en haut et au fond de cuve pour le contrôle de l’aspect.
Contrôles et critères d’acceptation
Tableau 5 : IPC au cours de la préparation de la phase aqueuse
IPC Production/LCQ Spécifications
Evaluation visuelle Solution limpide
2.2.Préparation de la phase grasse
Objectif : Vérifier l’aspect du mélange.
Echantillonnage : Dans le mélangeur, après le temps d’agitation et d’homogénéisation
27 | P a g e
Contrôles et critères d’acceptation
Tableau 6 : IPC au cours de la préparation de la phase grasse
IPC Production/LCQ Spécifications
Evaluation visuelle Mélange homogène avec absence de grumeau
2.3.Préparation de la solution du principe actif
Objectif : Vérifier la dissolution du principe actif.
Echantillonnage : Dans le conge 20L, après le temps d’agitation de 20 min, prélever en haut et au fond du conge pour vérifier la dissolution du principe actif.
Contrôles et critères d’acceptation
Tableau 7 : IPC au cours de la préparation de la solution du principe actif
IPC Production/LCQ Spécifications
Evaluation visuelle Mélange limpide
2.4.Mélange final (Vrac)
Objectif : Vérifier les caractères pharmaco techniques de la crème (Aspect, pH), dosage de la substance active, du propyle gallate, de l’acide sorbique et du parahydroxybenzoate de méthyle.
Echantillonnage : Dans le mélangeur, après le temps d’agitation de 40 min, arrêter l’agitation et prélever :
3 échantillons de 200 g en haut, au milieu et en fond de cuve pour les caractères pharmaco techniques (9 points de prélèvement).
3 échantillons de 100 g en haut, au milieu et en fond de cuve pour la teneur de la substance active, de l’acide sorbique, du parahydroxybenzoate de méthyle, du propyle gallate, de l’eau et en matières extractibles (9 points de prélèvement).
Contrôles et critères d’acceptation
Tableau 8 : IPC durant la validation du mélange final
IPC Production/LCQ Spécifications
Aspect Crème blanche, pratiquement inodore, onctueuse
Contrôle de l’homogénéité Pas d’hétérogénéité visible sur le film de crème étalé entre deux plaques
pH 3,50-4,50 Dosage Substance active Acide sorbique Parahydroxybenzoate de méthyle Eau Matières extractibles Propyle gallate Individuelle : 85,0-115,0 mg/100g Moyenne : 96,0-104,0 mg/100g RSD ≤ 6% < 200,0 mg/100g RSD ≤ 6% < 157,5 mg/100g RSD ≤ 6% 59,4-65,7% 33,3-44,7% <10,0 mg/100g RSD ≤ 6%
28 | P a g e
2.5.Stockage
Objectif : Vérifier les caractères pharmaco-techniques de la crème (Aspect, pH), dosage de la substance active, de l’acide sorbique, du parahydroxybenzoate de méthyle, des substances apparentées, la teneur en eau, en matières extractibles et en propyle gallate.
Echantillonnage
Dans la cuve de stockage prélever :
1 échantillon de 200 g en haut, au milieu et en fond de cuve pour les caractères pharmaco techniques (2 points de prélèvement).
1 échantillon de 100 g en haut et en fond de cuve pour le dosage de la substance active, de l’acide sorbique, du parahydroxybenzoate de méthyle, de l’eau, des matières extractibles et du propyle gallate (2 points de prélèvements).
Contrôle et critères d’acceptation
Tableau 9 : IPC durant la validation du stockage
IPC Production/LCQ Spécifications
Aspect Crème blanche, pratiquement
inodore, onctueuse Ph 3,50-4,50 Dosage Substance active Acide sorbique Parahydroxybenzoate de méthyle Eau Matières extractibles Propyle gallate 95,0-105,0 mg/100g < 200,0 mg/100g < 10,0 mg/100g 59,4-65,7% 33,3-44,7% 10,0 mg/100g
3. Validation du procédé de répartition (conditionnement primaire)
Objectif : Vérifier que le procédé de remplissage conduit à un produit conditionné conforme aux spécifications. Les caractéristiques étant : Aspect, pH, dosage de la substance active, de l’acide sorbique, du Parahydroxybenzoate de méthyle, de l’eau, des matières extractibles, de propyle gallate et la propreté microbienne.
Echantillonnage : une fois les réglages effectués, procéder à l’échantillonnage. Par journée de répartition (tube)
Tableau 10 : échantillonnage pour la validation du procédé de répartition (Par journée de
répartition)
Contrôle Echantillon Fréquence (par jour)
Aspect 10 tubes Début, Milieu, Fin
29 | P a g e
Etanchéité 5 tubes
Uniformité de masse 20 tubes
Par lot (tube)
Tableau 11 : échantillonnage pour la validation du procédé de répartition (par lot)
Contrôle Echantillon Fréquence
pH 2 tubes Début, Milieu, Fin
Dosage de la substance active 2 tubes Début, Milieu, Fin
Teneur en eau 2 tubes Début, Milieu, Fin
Dosage de l’acide sorbique, le Parahydroxybenzoate de méthyle et du propyle gallate
3 tubes Début, Milieu, Fin
Propreté microbienne 4 tubes Début, Milieu, Fin
Contrôles et critères d’acceptation
Tableau 12 : Contrôle durant la validation du procédé de répartition
Contrôles Spécifications
Aspect Crème blanche, pratiquement inodore, onctueuse
Masse moyenne ≥ 15 g
Uniformité de masse 2 tubes au maximum < 15 g 0 tube inférieur < 14,7 g
Etanchéité L’étanchéité doit être à 0 défaut, toute autre
valeur entraîne un écartement de ce qui a été conditionné dans le temps écoulé entre deux tests. Ph 3,5 à 4,5 Dosage Substance active Acide sorbique Parahydroxybenzoate de méthyle Eau Matières extractibles Propyle gallate 95,0-105,0 mg/100g < 200,0 mg/100g < 157,5 mg/100g 59,4-65,7% 33,3-44,7% 10,0 mg/100g Propreté microbienne
Dénombrement des germes aérobies totaux.
Entérobactéries et certaines autres bactéries gram- Staphylococcus aureus Pseudomonas aeroginosa ≤100UFC/g <10 UFC/g Absence/1 g Absence/ 1g 4. Plan d’échantillonnage
30 | P a g e
Tableau 13 : plan d’échantillonnage pour la validation du procédé de fabrication de la crème Etape de
production
Opération Equipement Contrôle Echantillon Prélèvement Annexe du Plan de prélèvement
2 Préparation de la phase aqueuse
Cuve chauffante Aspect 2 Haut et Fond Annexe 1 3 Préparation de la
phase grasse
Mélangeur Aspect 2 Haut et Fond Annexe 1 5 Préparation de la
solution du principe actif
Conge 20L Aspect 2 Haut et Fond Annexe 1
6 Mélange final Mélangeur Caractères pharmaco techniques
9 x 200g Haut, Milieu et Fond
Annexe 1
Dosages 9 x 100g Haut, Milieu et Fond 7 Stockage Cuve de stockage Caractères
pharmaco techniques
2 x 200g Haut et Fond Annexe 1
Dosages 2 x 100g Haut et Fond 8
Répartition Remplisseuse
Aspect 10 tubes Début/Milieu /Fin (par jour) Masse moyenne Uniformité de masse 20 tubes Etanchéité 5 tubes pH 2 tubes
Dosages 7 tubes Début-Milieu-Fin
(Par lot) Propreté
microbienne
4 tubes
5. Validation du conditionnement primaire et secondaire
Objectif : Vérifier sur un échantillonnage statistique du lot que des boîtes produites sont sans défauts apparents et que la remplisseuse produit des tubes sans défauts. Les défauts rencontrés sont classés en défauts rédhibitoire (R), critique (C), majeur (M) et mineur (m). Echantillonnage : L’échantillonnage est réparti sur l’ensemble du lot.
Le nombre d’échantillons prélevés doit être défini statistiquement selon une table statistique (Military Standard : Annexe 2). La table comprend 3 niveaux de contrôle : Niveau I (normal), Niveau II (allégé), Niveau III (renforcé). Dans le cadre de la validation, un contrôle renforcé est appliqué.
Selon la table statistique, un échantillon de 315 unité pour les contrôles du conditionnement secondaire et primaire.
Contrôle
31 | P a g e
Les défauts sont comptabilisés par nature rédhibitoire (R) critique (C), majeur (M) ou mineur
(m).
Tableau 14 : table de décision des défauts du conditionnement secondaire
Défaut rédhibitoire Défaut critique Défaut majeur Défaut mineur
-Etui étranger ou absent (R) -Notice étrangère ou absente (R) -Article de conditionnement non conforme (R)
-Mentions mobiles non conformes ou illisibles (R) -Contenant vide (R) -Unité étrangère (R)
-Etuis vides ou incomplets (C)
-Notice mal coupée avec perte d’une partie du texte (C)
-Notice déchirée (M) -Etui abîmé, fortement taché (M)
-Mentions mobiles décalées (M)
-Mentions mobiles estompées mais lisibles (M)
-Notice mal pliée, froissée (m)
-Notice mal coupée mais lisible (m) -Notice/étui légèrement tâché (m) -Notice supplémentaire (m) -Marquage imparfait mais lisible (m) Critères d’acceptation
Tableau 15 : Niveau de qualité acceptable des défauts du conditionnement secondaire Rédhibitoire (0 défaut) Critique (NQA 0,15) Majeur (NQA 0,65) Mineur (NQA 2,5)
Accepté 0 0 boîte 3 boîtes 12 boîtes Refusé 0 1 boîte 4 boîtes 13 boîtes
Contrôle du conditionnement primaire Prendre 315 boîtes représentatives du lot soit 315 tubes.
Comptabiliser les défauts par nature rédhibitoire (R), critique (C), majeur (M), mineur (m).
Tableau 16 : table de décision des défauts du conditionnement secondaire
Défaut rédhibitoire Défaut critique Défaut majeur Défaut mineur
-Unité étrangère (R) -Tube vide (R) -Corps étranger (R) -Mauvais article de conditionnement (R) -Mentions mobiles non-conformes (R)
-Tube incomplet (C) -Mauvaise étanchéité du tube (C)
-Unité écrasée, cassée (C)
-Tache, salissure à l’intérieur de l’unité (C) -Marquage absent ou illisible (C)
-Unité altérée, souillée, tachée (M)
-Mentions mobiles décalées (M)
-Mentions mobiles estompées mais lisibles (M)
-Taches, salissures (m)
-Autres défauts esthétiques (m)
32 | P a g e
Critères d’acceptation
Tableau 17 : Niveau de qualité acceptable des défauts du conditionnement primaire Rédhibitoire (0 défaut) Critique (NQA 0,15) Majeur (NQA 0,65) Mineur (NQA 2,5)
Accepté 0 0 tube 3 tubes 12 tubes Refusé 0 1 tube 4 tubes 13 ubes
IV.
Résultats et discussion
Dans le but de maitriser le procédé de fabrication de la crème tube de 15 g, une démarche de validation a été entreprise suite aux changements de la taille de lot et d’équipement. Une validation prospective est entreprise sur 3 lots.
1. Description du procédé de fabrication
La taille de lot mise en œuvre est : 429 Kg soit 28600 tubes de 15g. a. Schéma du procédé de fabrication
Opérations Etapes Matériel
Fusion 1 Ceinture chauffante
Température = 200°C Température MP = 70°C
Préparation de la phase aqueuse 2 Cuve chauffante Double paroi 350L
Température= 73 à 77°C Vitesse d’agitation=5 Durée d’agitation=30 min
Préparation de la phase grasse 3 Mélangeur
Vitesse d’homogénéisation = vit I Vitesse d’agitation= 15 tr/min
Durée d’agitation=15 min Température= 73 à 77°C
Emulsification 4 Mélangeur
Température phase aqueuse = 73 à 77 °C Température phase grasse = 73 à 77 °C
Vitesse d’agitation = 25 tr/min Durée =15 minutes
Vide =-0.5 bar Refroidissement= 50°C
Préparation de la solution de principe actif
5 CONGE INOX 20 L
Température de chauffage= 45 °C Durée =20 minutes Maintenir la température à 50°C
33 | P a g e
Mélange final 6 Mélangeur
Vitesse d'agitation= 40 tr/min Dépression= -0,5 bar Durée de mélange= 40 à 48 min
Température= 30°C pH = 3,5 à 4,5
Stockage 7 Cuve de stockage
Vitesse d'agitation = 25 tr/min Durée d'agitation= 30 min
Température= 32°C
Répartition 8 Remplisseuse, Mth = 15 g
Conditionnement secondaire 9 Encartonneuse
Figure 9 : Schéma du Process de fabrication de la crème avec les paramètres du contrôle
2. Validation du procédé de fabrication
a. Préparation de la phase aqueuse (étape 2)
L’aspect de la solution des deux niveaux haut et fond de la cuve des trois lots de validation est limpide. L’évaluation visuelle des trois lots de validation satisfait aux critères d’acceptation.
b. Préparation de la phase grasse (étape 3)
L’aspect du mélange des deux niveaux haut et fond de la cuve des trois lots de validation est homogène avec absence de grumeau. L’évaluation visuelle des trois lots de validation satisfait aux critères d’acceptation.
c. Préparation de la solution du principe actif (étape 5)
La solution du principe actif des deux niveaux haut et fond de la cuve des trois lots de validation est limpide. L’évaluation visuelle des trois lots de validation satisfait aux critères d’acceptation.
d. Mélange final (étape 6) 1. Aspect
L’aspect des trois niveaux des trois lots de validation est conforme aux spécifications. Absence de l’hétérogénéité visible sur le film de crème étalé entre deux plaques de verre.
2. pH
Tableau 18 : Résultats du contrôle du pH
Lot MA001V Lot MA002V Lot MA003V Spécifications
Haut cuve 3,6 3,7 3,7
3,5 à 4,5
Milieu cuve 3,6 3,7 3,7
Fond cuve 3,6 3,7 3,7
Le pH des trois niveaux des trois lots de validation est conforme aux spécifications. Le RSD des différents points des trois lots de validation est 1,4%, montrant une bonne homogénéité du mélange.
34 | P a g e
Tableau 19 : Résultats du contrôle de la teneur en eau
Lot MA001V Lot MA002V Lot MA003V Inter -lot Normes 1 Ch1/4 63,7% 64,7% 64,0% 59,4 à 65,7% 1 Cm1/4 64,2% 62,1% 63,4% 1 Cf1/4 63,1% 59,4% 62,9% 1 Ch1/2 63,7% 62,2% 62,2% 1 Cm1/2 64,5% 62,3% 61,7% 1 Cf½ 63,4% 64,8% 62,6% 1 Ch¾ 63,9% 63,1% 64,0% 1 Cm ¾ 63,7% 62,1% 62,1% 1 Cf¾ 63,6% 64,6% 62,1% Moyenne 63,76% 62,81% 62,78% 63,11% RSD 0,7% 2,8% 1,3% 0,7% ≤ 6,0% La teneur en eau des neufs niveaux des trois lots de validation est conforme aux spécifications.
Capabilité
Figure 9 : Capabilité du procédé de fabrication-Teneur en eau
La figure montre que la distribution est dans les limites de spécification. La moyenne +3ϭ = 66,68
La moyenne -3ϭ = 59,54
Les résultats sont situés dans l’intervalle de tolérance Moy ± 3σ. La Moy +3σ est supérieure à la limite de spécification supérieure qui peut être justifié par le fait que la teneur en eau est la somme de la teneur en eau ajoutée et la teneur en eau intrinsèque de la matière première. On peut conclure alors que la répartition de l’eau dans le mélange est homogène.
35 | P a g e
Tableau 20 : Résultats du contrôle de la teneur en acide sorbique
Lot MA001V Lot MA002V Lot MA003V Inter –lot Spécifications 1 Ch1/4 187,8 192,6 192,7 < 200,0 mg/100g 1 Cm1/4 189,2 195,9 192,6 1 Cf1/4 192,2 196,2 191,5 1 Ch1/2 188,9 195,2 191,7 1 Cm1/2 189,4 192,1 191,9 1 Cf½ 187,9 194,3 190,1 1 Ch¾ 189,3 194,8 189,9 1 Cm ¾ 186,5 193,3 191,2 1 Cf¾ 190,1 191,8 191,0 Moyenne 189,0 194,0 191,4 191,5 RSD 0,8% 0,8% 0,5% 1,3% ≤ 6,0% La teneur en Acide sorbique des neufs niveaux des trois lots de validation est conforme aux spécifications. La répartition de l’Acide sorbique dans le mélange est homogène.
Capabilité :
Figure 10 : Capabilité du procédé de fabrication-teneur en acide sorbique
La figure montre que la distribution est dans les limites de spécification. La moyenne +3ϭ = 198,97
La moyenne -3ϭ = 183,99
Les résultats sont situés dans l’intervalle de tolérance Moy ± 3σ. La répartition de l’Acide sorbique dans le mélange est homogène et satisfaisante.