Development of Polymer - Lipid Nanoparticles for Potent mRNA
Delivery to the Lung
By
James Cliff Kaczmarek Bachelor of Chemical Engineering
Auburn University, 2013 M.S. Chemical Engineering Practice Massachusetts Institute of Technology, 2015
SUBMITTED TO THE DEPARTMENT OF CHEMICAL ENGINEERING IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY AT THE
MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER 2018
2018 Massachusetts Institute of Technology. All rights reserved.
Signature of Author:
Signature redacted
James C. Kaczmarek Department of Chemical Engineering July 19, 2018
Signature redacted
Certified by:
V'
Associate Professor of Chemical Engineering~~~~~~Oaniel G. Anderson Thesis SupervisorAccepted by:MASSACHUSET1S INSTITUTE OF TECHNOLOGY
MAY
0
2Z1j
LIBRARIES
Siqnature redacted
Patrick S. Doyle Robert T. Haslam (1911) Professor of Chemical Engineering
Development of Polymer - Lipid Nanoparticles for Potent mRNA Delivery to the Lung By
James C. Kaczmarek
Submitted to the Department of Chemical Engineering on July 1 9th, 2018 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
ABSTRACT
Messenger RNAs (mRNAs) are an emerging therapeutic modality that holds great promise to specifically and completely treat genetic disease. mRNA has been used as a vaccine, as a protein replacement therapy, and even as a means of inducing permanent genomic editing via CRISPR. However, unlike traditional small molecule drugs, naked mRNA cannot readily enter the cellular cytosol, where it must localize in order to be successfully translated. The field of nucleic acid delivery, therefore, is largely concerned with the development of materials which can encapsulate mRNA and facilitate its transport into cellular cytoplasm in vivo. Many lipid nanoparticles originally designed to deliver short interfering RNA (siRNA) have been successfully repurposed to deliver mRNA, although their application is limited mainly to the liver. Thus, there is a continued need for the development of new materials for mRNA delivery in order to expand its therapeutic potential throughout the body.
Another class of nucleic acid delivery materials, poly(P-amino ester)s, or PBAEs, have been successful in delivering DNA cargo in vitro and in vivo, but their capacity for mRNA delivery has been relatively understudied. In this thesis, we utilized formulation techniques developed for lipid nanoparticles to systemically deliver mRNA-loaded PBAEs. We showed that non-covalent formulation of PBAE-mRNA nanoparticles with a polyethylene glycol-lipid conjugate imparts serum stability to the nanoparticles, which in turn correlates with in vivo efficacy. Specifically, we demonstrated that these materials are mainly effective in lung tissue following intravenous administration. The lung targeting and potency of these nanoparticles was then greatly improved through statistical optimization of the polymer synthesis and nanoparticle formulation. These optimized nanoparticles transfected the majority of lung endothelial cells, as well as a variety of immune cells populations. The nanoparticles were also used as a means of quantitatively comparing mRNA and DNA delivery in vitro and in vivo. We showed a drastic decrease in DNA potency in vivo compared to mRNA, attributed to the difficulty in crossing the nucleus of slowly dividing cells. Moreover, we observed similar kinetics of protein expression between mRNA and DNA. Additionally, we demonstrated in in vitro proof-of-concept studies that PBAE nanoparticles are capable of CRISPR-mediated genome editing.
We also show successful mRNA delivery in a variety of other tissues beyond the lung endothelium. Through the development of novel chemistries using both PBAEs and lipids, we were able to achieve mRNA delivery specifically to the lung endothelium as well as the spleen in
vivo. Taken together, the materials developed herein greatly expand the therapeutic capabilities
of mRNA. It is our hope that the work presented in this thesis translates into therapeutically relevant treatments while also providing insight into design criteria for successful mRNA delivery.
Thesis Supervisor: Daniel G. Anderson Title: Associate Professor
ACKNOWLEDGEMENTS
As with all other undertakings of any sort of significance, this work would not have been possible without the help of a host of others, only a fraction of whom I have room on this page to mention. First, I have to thank my advisor, Prof. Daniel G. Anderson, for giving me the opportunity to work as a part of his lab. The freedom and support I had to pursue open-ended scientific questions, while sometimes daunting, helped me to grow tremendously as a researcher. I would also like to thank the other members of my thesis committee, Profs. Robert Langer and Matthias Nahrendorf, for their support and helpful advice over the years. Thanks are also in order to Prof. Mark Byrne, my undergraduate mentor, who has been an incredible supporter and my inspiration for pursuing my PhD.
In addition to my thesis committee, I have also been incredibly blessed by a number of mentors in the form of post docs within the Anderson and Langer labs. Gaurav Sahay, Matt Webber, Sid Jhunjhunwala, and Asha Patel, in particular, were instrumental in my development as a scientist. Without the four of them, I would have been lost... and probably still contaminating my cells. I cannot thank them enough for investing time in me, and I hope the work herein in some small way justifies their efforts on my behalf.
For all but my final year in the lab, I had the extraordinary opportunity to follow in the footsteps of my colleague, co-author, and one of my best friends, Kevin Kauffman. I cannot thank Kevin enough for always being there to teach me, to listen to me, and to show me what being an honest, hard-working researcher looks like. Additional thanks are in order to Owen Fenton, without whom I would know substantially less chemistry and would have had substantially less fun in lab. I also want to thank all the other current and former graduate students in the lab, especially Luke Rhym, Abel Cortinas, Umberto Palmiero, Lisa Volpatti, Hok Hei Tam, and Amanda Facklam for making the lab a fun and welcoming place to work.
Since our first semester together, I have treasured the unique bond I've been able to make with my classmates, a bond that has made even the toughest times more bearable. So, to each and every member of the Course X graduate students of 2013, thank you for being my friend and keeping me sane. A special shout out is in order for my practice school group, as well as my practice school mentors, Bob Hanlon and Bob Fisher, who truly helped me to get the most out of the Practice School experience. And to that one special group of intrepid adventurers, Harry Watson, Carlos Pons Siepermann, Zsigi Varga, Andrew Fiore, Orpheus Chatzivasileiou, and Daniel Consoli, Randrok thanks you and looks forward to continuing our adventures, and continuing to ensure Domino's Pizza stays in business.
The greater Boston area is the farthest I have ever lived from my biological family, however the support I've received these past 5 years from my church family has truly made this area feel like home, blizzards and all. So thank you, Hope Fellowship Church, for providing me with all the love and support I could ever need, and then some. Of course, distance has not stopped my biological family from supporting me with all that they have. Thanks, Mom and Dad, for always supporting in me and believing in me. All that I've been able to accomplish has been made possible by your love and sacrifice for my sake.
Of course, this section would be incomplete if I did not thank the love of my life. Anna Beth, getting to come home to you every evening makes even the most difficult days bearable. Thank you for laughing with me, watching anime with me, cheering for Auburn with me, and loving me despite my flaws. I love you, and could not imagine completing this thesis, or living life, without you.
Finally, I ascribe any and all glory and honor associated with this thesis to God, in whose Name this work has been completed.
TABLE OF CONTENTS
CHAPTER 1 - INTRODUCTION AND BACKGROUND ... 9
1 .1 M o tiv a tio n ... 1 0 1.1.1 Materials and Chemical Modifications for Nucleic Acid Delivery ... 11
1.1.2 mRNA Delivery... 15
1.1.3 mRNA Delivery for Gene Editing via CRISPR/Cas9 ... 20
1.2 Nucleic Acid Delivery using PBAEs...24
1.2.1 Combinatorial Library Synthesis of PBAEs for Gene Delivery...25
1.2.2 Non-Combinatorial Library Approaches for PBAEs... 33
1. 2.3 Other Uses for PBAEs Es... ... 36
1.3 Perspective and Thesis Overview ... 40
CHAPTER 2 - FORMULATING POLY(B-AMINO ESTER) NANOPARTICLES FOR SYSTEMIC MRNA DELIVERY ... 42
2 .1 In tro d u c tio n ... 4 3 2.2 Polymer Synthesis and Formulation with mRNA ... 44
2.3 Correlation between in vitro efficacy and PBAE nanoparticle stability ... 45
2.4 Intravenous administration of PEGylated PBAE nanoparticles... 49
2 .5 C o n clu sio n s ... . . 5 2 2.6 Materials and Methods... 53
2.7 Acknowledgements...61
CHAPTER 3 - OPTIMIZATION OF PBAE NANOPARTICLES FOR SYSTEMIC MRNA DELIVERY TO THE LUNGS ... 62
3 .1 Intro d u ctio n ... . . 6 3 3.2 PBAE Nanoparticle Optimization... 64
3.2.1 Polymer Synthesis Optimization ... 64
3.2.2 Formulation Optimization...69
3.3 Batch Consistency of Optimized Polymers...74
3.4 Characterization of Lung Cell Types Transfected... 77
3.4.1 The Ail4 Cre-lox Mouse Model ... 77
3.4.2 Probing the Lung Cells Transfected with Optimized PBAEs ... 79
3 .5 C o n clus io n s ... . . 8 0 3.6 Materials and Methods... 81
CHAPTER 4 - USING OPTIMIZED PBAE CHEMISTRY TO COMPARE MRNA AND DNA
DELIVERY ... 91
4 .1 In tro d u c tio n ... 9 2
4 .2 R e s u lts ... 9 4
4.2. 1 In vitro Screens for DNA Efficacy... 94 4.2.2 In vivo Screen for DNA Efficacy... 96 4.2.3 Nanoparticle Characterization... 99 4 .3 D is c u s s io n ... 1 0 2
4 .4 C o n c lu s io n s ... 1 0 6
4.5 Materials and Methods...107 4.6 Acknowledgements...115
CHA PTER 5 - MRNA DELIVERY IN OTHER TISSUES... 116
5 .1 In tro d u c tio n ... 1 1 7 5.2 Nebulized Delivery of Branched PBAEs ... 118 5.3 Splenic Delivery Using Polycaprolactone-based PBAEs ... 121
5.4 Lipid Nanoparticles for Non-Lung m RNA Delivery...124
5 .5 C o n c lu s io n s ... 1 2 6 5.6 Materials and Methods...127
5.7 Acknowledgements...134
CHA PTER 6 - G ENE EDITING USING PBA ES ... 135
6 .1 In tro d u c tio n ... ... 1 3 6 6.2 Gene Editing in HEK Cells ... 137 6 .3 C o n c lu s io n s ... 1 3 9
6.4 Materials and Methods...140 CHA PTER 7 - CONCLUSIONS ... 143
7 .1 S u m m a ry ... 1 4 4 7.2 Future W ork ... 144
7.2.1 Application in a Therapeutic Model...144
7.2.2 Development of Next-Generation PBAE Chemistries ... 146 7.2.3 Understanding the Pharmacokinetics and Toxicity of PBAE Nanoparticles
146
LIST OF FIGURES
Figure 1-1: Common delivery modalities for RNA... 15
Figure 1-2: Regulation of gene and protein expression using RNA. ... 19
Figure 1-3: Number of new PBAE publications each year (data from MEDLINE). ... 25
Figure 1-4. PBAE and nanoparticle synthesis. ... 26
Figure 1-5. Alternate PBAE Syntheses... 29
Figure 1-6. Non-nucleic acid delivery uses for PBAEs... 37
Figure 2-1: Representative synthesis of PBAE terpolymers. ... 44
Figure 2-2: PBAE polymer synthesis and nanoparticle formulation. ... 45
Figure 2-3: PBAE serum stability and in vitro efficacy. ... 48
Figure 2-4: Rescuing PBAE serum stability with increasing PEG-lipid... 49
Figure 2-5: In vivo efficacy of PBAE nanoparticles ... 52
Figure 3-1: Materials for optimization of PBAE nanoparticles... 65
Figure 3-2: Results of PBAE synthesis optimization... 68
Figure 3-3: Optimizing PBAE formulation for mRNA delivery to the lung. ... 73
Figure 3-4: Batch-to-batch variability of Al polymer. ... 75
Figure 3-5: SEC-based purification of Al PBAE polymer. ... 76
Figure 3-6: Using the Ai14 Cre-lox mouse model for single cell transfection analysis... 79
Figure 3-7 Analysis of lung cells transfected using A1-L3 nanoparticles... 80
Figure 4-1: Synthesis of PBAEs and PBAE nanoparticles... 95
Figure 4-2: In vitro transfections with mRNA and DNA. ... 96
Figure 4-3: In vivo screen for DNA efficacy. ... 98
Figure 4-4: Time course expression studies in vivo... 99
Figure 4-5: Nanoparticle physical characterization. ... 100
Figure 4-6: Nanoparticle endosomal trafficking studies. ... 102
Figure 5-1: Nebulization of mRNA-containing nanoparticles...119
Figure 5-2: Branched PBAEs for nebulized delivery to the lung epithelium. ... 121
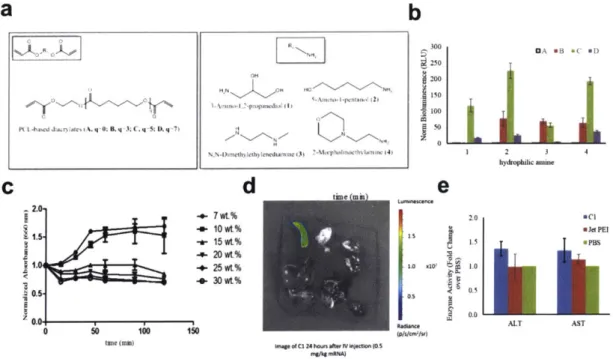
Figure 5-3: PCL diacrylate-based PBAEs for in vivo mRNA delivery. ... 124
Figure 5-4: Lipid nanoparticles for mRNA delivery. ... 126
Figure 6-1: PBAE-based CRISPR gene editing...138
Figure 6-2: Gene editing using PBAEs in HEK 293T cells...139
Figure A.1-1: 1H NMR, IR spectrum, GPC Trace, and Structure of DD24-C12-122...152
Figure A.1-2: 1H NMR, IR spectrum, GPC Trace, and Structure of DD60-C12-122...153
Figure A.1-3: 1H NMR, IR spectrum, GPC Trace, and Structure of DD90-C12-122...154
Figure A.1-4: Optimization of PEG-lipid amount...155
Figure A.1-5: Additional cryoTEM images of DD90-C12-122 with 7 mol% PEG-lipid...155
Figure A .1-6 : D ialysis at different pH ... 156
Figure A.1-7: Changes in concentration following serum stability assay...156
Figure A.1-8: Intravenous injection of highly PEGylated PBAE. ... 157
Figure A.1-9: PEGylated DD90-C12-122 biodistribution...157
Figure A.2-1: Characterization of Al polymer...161
Figure A.2-2: Model results for synthesis screen...161
Figure A.2-3: Model results for formulation definitive screen. ... 162
Figure A.2-4: Model results for formulation partial factorial screen (lungs). ... 162
Figure A.2-5: Model results for formulation partial factorial screen (spleen). ... 163
Figure A.2-6: Model results for formulation partial factorial screen (particle diameter)...163
Figure A.2-7: Correlation between diameter and luciferase signal for the partial factorial fo rm u la tio n s c re e n . ... 16 4 Figure A.2-8. Diameters of particles from the PEG-lipid screen...164
Figure A.2-9: Comparison of microfluidic mixing and pipette mixing...165
Figure A.2-1 0: Correlation between particle diameter and weight loss in mice at 24 hours for the partial factorial form ulation screen...165
Figure A .2-1 1: A 1-L 3 toxicity evaluation...166
Figure A.2-12: Biodistribution of mRNA translation. ... 167
Figure A.2-13: Comparison of mRNA delivery by lipid and PBAE nanoparticles...168
Figure A.2-14: SEC trace for Al PBAEs...168
F ig u re A .2-15 : L u ng ce ll g ating . ... 16 9 Figure A.2-16: Gating strategy for identifying lung immune cell populations...170
Figure A.2-17. Gating for tdTomato positive immune cells (x axis)...171
Figure A.3-1: Representative synthesis of DD90-C12-103 terpolymer. ... 172
Figure A.3-2: Residual plot of the linear regression comparing the mRNA and DNA efficacy in H e L a c e lls .... 1 7 2 Figure A.3-3: Identificaiton of D90-C12-103-transfected cell types...173
Figure A.3-4: Size control of DD90-C12-103...173
Figure A.3-5: Comparison of mRNA and DNA efficacy in D90-C12-103 and DD90-C12-103 with a nd w ith o ut thym id ine b lo ck. ... 174
Figure A.3-6: Cell cycle analysis of thymidine-treated HeLa cells...175
F ig u re A .3-7 : G ating S trateg y ... 17 6 Figure A.3-8: Comparison of tdTomato+ signal in lung cells for both treated and control mice. ... 1 7 6 Figure A.4-1: PCL-PBAE serum stability...176
Figure A.4-2: PCL diacrylate-based PBAEs vs. in vivo jetPEl. ... 177
LIST OF TABLES
Table 1-1 Comparison of clinically-relevant RNA delivery platforms...13
Table 1-2. Therapeutic applications of PBAEs delivering nucleic acids. ... 36
Table 5-1: Molecular weights of common diacrylates and amines for PBAE synthesis...122
Table A.1-1: Terpolymer nanoparticle characterization. ... 151
Table A.2-1: Parameter range for synthesis screen. ... 158
Table A.2-2: Conditions for synthesis screen. ... 158
Table A.2-3: Parameter ranges for definitive screen...159
Table A.2-4: Conditions for definitive formulation screen...159
Table A.2-5: Conditions for partial factorial formulation screen...159
Table A.2-6: Conditions for PEG-lipid formulation screen...160
Table A.2-7: Antibodies used in FACS analysis. ... 160
CHAPTER 1
- INTRODUCTION AND BACKGROUNDPortions of the work presented in this chapter were published as:
Kaczmarek, J.C., Kowalski, P.S., Anderson, D.G. "Advances in the Delivery of RNA Therapeutics: From Concept to Clinical Reality." Genome Medicine, 9(1), 60, 2017.
Kaczmarek, J.C., Patel, A.K., Anderson, D.G. "Poly(P-amino esters): Versatile Chemistry for a Variety of Drug Delivery Applications." In Preparation.
1.1 Motivation
14 years after the completion of the human genome project, our understanding of human genomics continues to develop at an unprecedented rate. Thanks to advances in next generation sequencing technology, scientists have been able to identify the genetic roots of many common diseases.' Diseases such as cancer,2 Parkinson's,3 rheumatoid arthritis,4 and
Alzheimer's' have all had many of their genetic components revealed, bringing us closer than ever to personalized medicine.' Thus far, this knowledge has been well adapted for diagnostic use but has not yet been fully translated to pharmaceutical interventions addressing genetic defects underlying diseases. Currently, the two major structural classes of FDA-approved drugs are small molecules and proteins.7 Small molecule drugs, which consist predominantly of
hydrophobic organic compounds, typically act by deactivating or inhibiting target proteins through competitive binding. However, the proteins which may possess such binding pockets have been estimated to account for only 2-5% of the protein-coding human genome.8
Protein-based drugs, on the other hand, can bind with high specificity to a variety of targets (e.g. antibodies), or be used to replace mutated or missing proteins (e.g. delivering insulin for diabetes). Still, protein size and stability limit their utility towards many potential disease targets.7 True realization of the therapeutic potential of personalized genomics requires treatments beyond those offered by current small molecule and protein therapy.
In summation, both protein and small molecule drugs are limited in that they cannot target every disease-relevant protein or gene. The mRNA and DNA precursors of proteins, however, are promising therapeutically in that they can be specifically targeted via Watson-Crick base pairing and, in the case of gene editing, which aims to permanently change the host's DNA, represent an avenue to cure a genetic defect as opposed to just treating it. Over the past few decades, nucleic acid drugs, especially those based on RNA, have emerged as candidates to address diseases on a gene and RNA level. Although it has been known since 1990 that
nucleic acids can be used to modulate protein production in vivo,9 therapeutic RNA delivery has
been limited by a number of factors. Naked, single-stranded RNA is prone to nuclease degradation, can activate the immune system, and is too large and negatively charged to passively cross the cell membrane and must therefore possess additional means of cellular entry and endosomal escape.10 As such, the RNA delivery field has centered on the design of delivery methods and materials that will transport RNA drugs to the site of interest.
1.1.1 Materials and Chemical Modifications for Nucleic Acid Delivery
To summarize the previous points, an ideal nucleic acid delivery vehicle will
1) Protect the nucleic acid cargo from degradation by nucleases
2) Facilitate cellular uptake, nuclear localization (in the case of DNA), and endosomal escape of the nucleic acid cargo
3) Demonstrate appropriate stability, efficacy, and biodistribution in vivo
4) Demonstrate low levels of toxicity and immunogenic response
In order to accomplish the above, many materials have been developed for nucleic acid delivery. Broadly speaking, nucleic delivery can be mediated by viral and non-viral vectors, and many of the factors that are important for RNA delivery are also relevant for DNA delivery. For viral RNA delivery, there has been a great deal of interest in engineering adeno-associated viruses to carry nucleic acid cargo," however this thesis will focus on the development of non-viral materials. Of the non-non-viral RNA delivery vehicles, nanoparticles are perhaps the most-studied. Nanoparticle encapsulation of RNA physically protects nucleic acids from degradation and, depending on the specific chemistry, can aid in cellular uptake and endosomal escape. Given their high degree of chemical flexibility, polymers are commonly used materials for nanoparticle-based delivery.1 2 Typically, cationic polymers are used to electrostatically
condense the negatively-charged RNA into nanoparticles (Figure 1-1a).13 These
positively-charged groups often consist of amines which become protonated at physiological pH (pKa -7.4), thought to lead to an ion imbalance that results in endosomal rupture, 14,15 although this
so-called "proton sponge" hypothesis has yet to be rigorously demonstrated for various materials.16 Synthetic polymers such as poly-L-lysine,17 polyamidoamine,18 and
polyethyleneimine (PEI),19 as well as naturally-occurring polymers such as chitosan20 have all
been applied to RNA delivery with varying levels of success. In addition, some groups have synthesized polymers specifically for nucleic acid delivery. Poly(P-amino esters), in particular, have gained widespread use in DNA delivery due to their ease of synthesis and biodegradability21 but have also proved to be capable of short interfering RNA (siRNA)2 2
-2 4 and
mRNA25 delivery.
Lipids and lipid-like materials represent the second major class of nanoparticle-based delivery vehicles for RNA. As with polymers, cationic lipids are often used in order to electrostatically bind the nucleic acid. Many groups, however, have started utilizing ionizable lipids, which are lipids that are positively charged only at acidic pH. This ionizable behavior is thought to enhance efficacy through helping with endosomal escape and reducing toxiCity27 as compared to cationic particles. Lipids are also capable of self-assembly into well-ordered nanoparticle structures, known as lipoplexes (Figure 1-1b), driven by a combination of electrostatic interactions with RNA and hydrophobic interactions.2 2,9 Optimizing the formulation
of lipid nanoparticles by addition of other hydrophobic moieties, such as cholesterol and PEG-lipid, in addition to an ionizable/cationic PEG-lipid, enhances nanoparticle stability and can significantly enhance efficacy of RNA delivery.30 However, similarly to polymers, it was found that ionizable lipid structure is the main factor affecting efficacy of the nanoparticle. As such, our lab pioneered the use of semi-automated high throughput synthesis methods to create libraries of chemically diverse lipids and lipid-like materials for RNA delivery, 31-3 resulting in highly
potent nanoparticles capable of delivering a variety of RNA types to both the liver32
,36,37 and the lung33 following systemic delivery in vivo.
As an alternative to nanoparticles, a more conceptually straightforward and chemically well-defined mean of delivery is to directly conjugate a bioactive ligand to the RNA that will allow it to enter the cell of interest. Perhaps the most clinically-advanced example of this technique is the conjugation of n-acetylgalactosamine (GaINAc, Figure 1-1c), which targets the asialoglycoprotein receptor on hepatocytes, to siRNA.38 Unlike many nanoparticles, which are given intravenously, GaINAc conjugates are typically dosed subcutaneously and have shown an ability to rapidly enter systemic circulation and target the liver.39 Other conjugates, such as cholesterol,4 0 vitamin E,41 antibodies,4 2 and cell-penetrating peptides43 have been explored in
the past, although none but the specialized triatennary GalNAc-siRNA conjugate has gained any clinical traction, suggesting the need for additional work on the design of conjugates for efficient nucleic acid delivery.
Table 1-1 Comparison of clinically-relevant RNA delivery platforms.
Delivery Vehicle Type of RNA in Advantages Disadvantages
Clinical Trials
Naked RNA siRNA, ASO, - No additional materials or - Prone to degradation
mRNA synthesis required - Immunogenic
- Difficulty entering cell
- Poor circulation
half-life
Nanoparticle siRNA, ASO, - Increased half life - Elevated risk of toxicity
mRNA - Protection from nucleases with introducing
- Aids in endocytosis and excipient materials
endosomal escape
Conjugate siRNA, ASO - Defined chemical structure - High doses required
- Ability to target specific - Dependent on
receptors chemical modifications
- Limited toxicity due to lack for RNA stability
of excipient materials
Also important for effective nucleic acid delivery are chemical modifications made to the RNA itself, which can impart degradation resistance to the RNA" and render them unrecognizable by the immune system. This is true of both conjugate delivery systems, which
leave the RNA exposed immediately upon injection, as well as nanoparticulate delivery systems, which must at some point expose the RNA to intracellular immune receptors. RNAs can be modified via chemical alterations to the ribose sugar (of particular importance is the 2' position45
,
46), the phosphate linkage, and the individual bases (Figure 1-1d).47
-50 RNAs delivered
via nanoparticle are also typically modified in order to avoid recognition by endosomally-expressed pattern recognition receptors.51 With few exceptions, modified RNAs are the gold
standard in clinical trials. The degree to which the RNA can be modified and still retain its potency depends, to a large extent, on the nature of the nucleic acid and its mechanism of action. For instance, short RNAs like siRNAs, which rely on the relatively robust RNA-induced silencing complex,2 can typically be heavily modified. Large mRNAs, which must be effectively
translated by ribosomes, are more sensitive to modifications and utilize naturally occurring RNA modification such as pseudouridine and 5-methylctidine substitution.53 Indeed, recent studies
(discussed in Chapter 5) have shown that base modification of mRNA may actually decrease potency in certain situations,5 4 whereas chemical modification in siRNAs is almost ubiquitously applied for in vivo use.55
b)
RNA Cargo
Catlonic Polymer RNA Cargo
Cationlc/lonizable Upid
C Phosphollpld
H0 O
AcHN HO~0 0 90O
AcHN 0 0 0 0 0 L _ N "OH OH OH H 0 HO O NN% AcHN o o
d )
B0s0 SONH, Bae HN NH B N 0 N o H0 2 HN N PHdHn B d H HH H0 H-Pseudouridine 5-Bromo-uridine S-metyictilne 2'-Oeoxy t-oeml3lnae Th
Chole
PEG esterol
boate inkage
Figure 1-1: Common delivery modalities for RNA. (a) Schematic depicting polymeric nanoparticles
composed of RNA and cationic polymer. (b) Schematic depicting lipid nanoparticles containing RNA, a cationic/ionizable lipid, and other hydrophobic moieties commonly used in nanoparticle formulation. (c) Chemical structure of the tertiary GaINAc-RNA conjugate currently in clinical trials. (d) Examples of base, sugar, and linker modifications that have been utilized to deliver nucleic acids (modified chemistry highlighted in blue).
1.1.2 mRNA Delivery
Expression of disease relevant proteins can be achieved by intracellular delivery of plasmid DNA (pDNA) or messenger RNA (mRNA). Application of a defined genetic message as a protein intermediate enables expression of virtually any desired protein inside the host cells and tissues. This modular approach can address formulation and delivery challenges encountered with protein-based drugs, especially those aimed at intracellular targets.6 mRNA-based therapeutics in particular offer several advantages over pDNA including rapid and
transient protein production, no risk of insertional mutagenesis and greater efficacy of non-viral delivery by virtue of mRNA cytoplasmic activity (Figure 1-2).11 Since the first pre-clinical studies in 1990s, mRNA technology has greatly developed and now holds a potential to revolutionize vaccination, protein replacement therapies and treatment of genetic diseases, consequently
gaining a considerable interest among the scientific community and biotech industry.53
The delivery of mRNA therapeutics has been enabled by significant progress in maximizing translation and stability of mRNA, preventing its immune-stimulatory activity and the development of in vivo delivery technologies, some of which are discussed subsequently. The 5' cap and 3' Poly(A) tail are the main contributors to efficient translation and prolonged half-life of mature eukaryotic mRNAs. Incorporation of Cap analogs such as ARCA (anti-reverse cap analogs) and Poly(A) tail of 120-150 bp into in vitro transcribed (IVT) mRNAs have markedly improved expression of encoded proteins and mRNA stability.58
.59 New types of Cap analogs
such as 1,2-dithiodiphosphate modified caps, with resistance against RNA decapping complex, can further improve RNA translation efficiency.60 Replacing rare codons within mRNA protein coding sequences with synonymous frequently occurring codons, so called codon optimization, also facilitates better efficacy of protein synthesis and limits mRNA destabilization by rare codons, thus preventing accelerated degradation of the transcript.61,62 Similarly, engineering 3'
and 5' untranslated regions (UTRs), that contain sequences responsible for recruiting RNA-binding proteins (RBPs) and miRNAs, can enhance the level of protein product.53,63 Interestingly, UTRs can be deliberately modified to encode regulatory elements (e.g. K-turn motifs and miRNA biding sites) providing means to control RNA expression in a cell specific manner.64 Previously discussed RNA base modifications such as N1-methyl-pseudouridine
have been instrumental not only in masking mRNA immune-stimulatory activity but were also shown to increase mRNA translation by enhancing translation initiation.65 66 In addition to their observed effect on protein translation, base modifications and codon optimization affect
secondary structure of mRNA, which in turn influences its translation.67 Understanding the importance and the ability to predict mRNA folding structure could aid engineering of mRNA therapeutics, however the accuracy of available prediction tools is currently limited. Despite the plethora of carriers studied for other types of RNA drugs, mRNA molecules are significantly larger (600-10,000 kDa) than siRNAs (-14 kDa) and ASOs (4-10 kDa), which poses an additional challenge for delivery of mRNA-therapeutic.68 Accommodation of large and charged mRNAs into nanoparticles and their effective intracellular release was shown to require fine tuning of existing formulations and the development of new generation of biomaterials with higher potency.6,37
Therapeutic applications of mRNA that are currently being explored are vaccinations against cancer and infectious disease, protein replacement therapy, and gene editing. mRNA vaccines are in the most advanced stages of clinical development, following in the footsteps of competing DNA and protein-based technologies. Synthetic mRNA vaccines allow simultaneous delivery of a wide variety of antigens and are both faster and easier to manufacture at low cost as compared with other systems, enabling more rapid response towards emerging pathogens.69 Additionally immune responses generated by naked mRNA can be beneficial for vaccination purposes.70
,71 Immunization against infectious diseases using ex vivo mRNA-transfected dendritic cells (DCs) is now pursued in clinical trials and demonstrated good safety profiles and ability to induce antigen specific T cell responses.72
Another RNA-vaccination approach is the use of self-amplifying mRNA replicons that have been developed to extend the duration and magnitude of antigen expression as well as boost the immune response.7374 In a recent study, replicon vaccines formulated into
nanoparticles composed of repeatedly branched dendrimer (tree-like) molecules have been able to generate protective immunity against a broad spectrum of lethal pathogen including Zika, Ebola and Influenza viruses.75 Conventional, modified mRNAs are also being explored for
vaccination.74 Lipid nanoparticle (LNP)-encapsulated mRNA encoding pre-membrane and
envelope glycoproteins of Zika virus have been recently reported to elicit potent and durable neutralizing antibody responses in mice and non-human primates against the virus after intradermal administration.76 Moreover, expression of modified mRNA encoding broadly
neutralizing antibody in the liver, after systemic administration of mRNA-LNPs, was able to protect humanized mice against HIV-1 challenge.77 Cancer mRNA-vaccines experienced
accelerated development and clinical translation driven by the success of cancer immunotherapy. The majority of approaches tested in clinical trials employ adoptive-transfer of DCs transfected with mRNAs coding for tumor specific-antigens (TCR) and immunomodulation of T cells with mRNAs expressing chimeric antigen receptors (CAR) or TCRs.78 In addition,
direct intradermal and systemic administration of LNP-formulated mRNAs coding for tumor-specific antigens is currently being investigated in the clinic for induction of T cell immune
responses.69,79,80 In contrast, most mRNA-based protein replacement therapies are still in the
preclinical stages of development and involve supplementation of deficient or aberrant proteins as well as modulation of cell behavior by expression of exogenous proteins. In vivo efficacy of RNA-protein therapy has been demonstrated for a number of diseases. The majority of the studies preferentially target the liver due to well established and efficient methods for RNA delivery into liver tissue. Therapeutically relevant amounts of human FIX (hFIX) protein were reached and sustained physiological activity for 4-9 days upon a single intravenous dose of hFIX mRNA-loaded LNPs in mice with hemophilia B.81,82 Similarly LNPs formulated with
Erythropoietin (Epo) mRNA were shown to elicit systemic physiological response in large animals including pigs and nonhuman primates. 2 Therapeutic effects of mRNA have been also
demonstrated in other tissues. Lung delivery of Surfactant protein B (SP-B) mRNA protected mice from respiratory failure, while myocardial injection of RNAiMAX formulated mRNA encoding human vascular endothelial growth factor-A (VEGF-A), improved heart regeneration after myocardial infarction in mice.84 Based on this notion, Astra Zeneca partnered with
Moderna therapeutics and launched a Phase I clinical trial for local delivery VEGF mRNA, starting January 2017 (www.modernatx.com). Pre-clinical studies demonstrated translational potential of mRNA-based protein therapy both for secreted and intracellular protein targets. However, treatment of chronic diseases may carry elevated risk of toxicity, associated with repeated mRNA-LNP administrations required to sustain therapeutic levels of protein. Using mRNA for delivery of gene editing tools could address this challenge and will be discussed below. mRNA Il"A. ASO I. siRNA siRNA .33 sgRNA IV. Ribosome >l % 1 ii Rnase H Cleaved mRNA
0
Protein 00 0 knockdown 00 Protein expression 0 Yvmu%~\ Cas9 RISCAM
0
000
00
4. IV CRISPR-Cas9 Cleaved DNA Protein knockout4-Figure 1-2: Regulation of gene and protein expression using RNA. Once delivered into the cells
RNA macromolecules can utilize diverse intracellular mechanisms to control gene and protein expression.
(1) Hybridization of antisense oligonucleotides (ASO) to a target mRNA can result in a specific inhibition of
gene expression by induction of RNase H endonuclease activity, which cleaves the mRNA-ASO
heteroduplex. (11) siRNA is recognized by the RNA-induced silencing complex (RISC), which guided by an
anti-sense strand of the siRNA specifically binds and cleaves target mRNA. (Ill) In vitro transcribed mRNA utilizes protein synthesis machinery of the host cells to translate the encoded genetic information into a protein. Ribosome subunits are recruited to mRNA together with a cap and poly(A)-binding proteins forming a translation initiation complex. (IV) Co-delivery of a single guide RNA (sgRNA) together with the mRNA coding for Cas9 protein allows site specific cleavage of double stranded DNA leading to a knockout of a target gene and its product.
1.1.3 mRNA Delivery for Gene Editing via CRISPR/Cas9
RNA-based technologies discussed in the previous sections constitute powerful means to transiently repress or overexpress function of genes. In contrast, therapeutic gene editing entails replacement or alteration of gene expression by introducing site-specific modifications into the genome of cells including correction of deleterious or introduction of protective mutations.5 While the majority of current gene editing efforts are focused on treatment of monogenic disorders, caused by deleterious changes in a single gene, the expansion of gene editing and delivery tools makes the treatment of complex polygenic diseases such as cardiovascular diseases6 and antiviral therapies,87 as well as editing the epigenome more feasible.88 The discovery of RNA-guided DNA endonucleases such as Cas9 associated with CRISPR (Clustered regularly interspaced short palindromic repeats), elements composing the prokaryotic adaptive immune system,89 equipped scientists with an easy-to-use and efficient
platform to alter genomic information.90 So-called CRISPR-Cas systems rely on Watson-Crick
base pairing between a single guide RNA (sgRNA) and a corresponding DNA target site followed by a distinct protospacer-adjacent motif (PAM), a 3-5 nucleotide DNA sequence required for binding of Cas9 and cleavage of the target sequence, to introduce a double stranded breaks (DSB) into a DNA.91 DSBs can be repaired by the cells using non-homologous end joining (NHEJ) and homology directed repair (HDR). NHEJ results in stochastic insertions and deletions (indels) causing permanent gene knockout, while HDR occurs in the presence of a DNA template containing homology to regions flanking DSB site, leading to incorporation of desired changes encoded in the repair template into the genome.92 A combination of DSBs can
also be used to edit multiple loci by employing different sgRNAs.93 94 To date the most widely
used and well characterized is the CRISPR-Cas9 system with an effector domain originating from Streptococcus pyogenes (SpCas9). Direct in vivo delivery of spCas9 to diseased cells has recently been used to correct mutations in animal models of Duchenne muscular dystrophy
(mdx),95-97 hereditary tyrosynemia type I (fah),98 99 lethal metabolic liver disease (oct),100 and to
reduce blood cholesterol in chimeric liver-humanized mice by knockout of PCSK9.101 Ex vivo
editing with spCas9 has been applied to human hematopoietic stem cells (HPSCs) in order to correct sickle cell anemia caused by mutation in
P-globin
gene, as well as to deplete T cells of CCR5 expression to trigger anti-HIV protection or PD-1 to boost anti-cancer therapy.102 Despitepositive outcomes, these studies revealed limitations of CRISPR-Cas9 system relevant for clinical translation including 1) imperfect DNA-targeting specificity leading to off-target effects 0 3
2) low efficiency of genome editing using HDR10 4 3) challenging delivery of CRISPR-Cas9
components using both viral and non-viral methods.105
DNA-targeting specificity of CRISPR-Cas9 can be improved by combining optimized design and synthesis of guide-RNAs. Namely, sgRNAs shorter than 20 nt and containing 5' mismatches have shown less off-target effects,106,107 while chemically synthesized sgRNAs
bearing base modifications at 5' and 3' ends demonstrated increased on-target efficacy.1 08
Furthermore, improved types of spCas9 such as high fidelity spCas9-HF1 109 or enhanced specificity eSpCas9110 have been engineered by introducing specific mutations into spCas9 based on interactions between spCas9-gRNA complex and DNA. New RNA-guided nucleases such as Cpfl from Acidaminococcus sp. (AsCpfl) with the capability to edit genome of mammalian cells have been recently discovered.1 '12 Cpfl nuclease mRNA (-1.3 kb) is
significantly smaller than Cas9, with different PAM requirement and inherently higher DNA-specificity than spCas9, which makes it attractive for clinical use. Off-target effects can be also limited by decreasing the cellular presence of spCas9 through conditions favoring transient over long-lasting expression, which can be accomplished by optimizing the delivery method.1081 1 3,
Obtaining better efficiency of genome editing by HDR will be necessary to address genetic diseases demanding a high level of therapeutic product, especially when edited cells do not display a positive change in fitness and outcompete their diseased counterparts over time.85
The efficiency of correction by HDR can be significantly improved by designing an asymmetric single stranded DNA template that anneals to the non-target DNA strand, which is the first to be released from Cas9-DNA complex.1 14 In addition, a number of studies reported
better HDR efficacy using CRISPR-Cas9 in combination with small molecule inhibitors of NHEJ e.g. DNA ligase IV or DNA-dependent protein kinase inhibitors. 115116 Alternatively, HDR can be
enhanced by agonists of proteins critically involved in homologous recombination such as Rad5l .117 Recently, other methods of gene editing with CRISPR-Cas9 called
homology-independent targeted integration (HITI) have been developed, which exploits NHEJ repair mechanism for gene knock-ins.1 HITI donor templates are designed to ensure robust gene integration only when inserted in the correct direction, otherwise target DNA would undergo additional cleavage by Cas9. This method demonstrated higher in vitro efficacy of transgene insertion as compared to HDR dependent editing but so far in vivo reached only 3-10 % knock-in efficiencies.
Intracellular delivery of CRISPR-based agents poses one of the most significant challenges for therapeutic genome editing due to the number of essential components. CRISPR-Cas9 components can be delivered as DNA, RNA, RNA-protein complex (RNP) or by combination of these macromolecules. These macromolecules are not able to spontaneously enter the cells, therefore rely on the use of delivery vehicles such as viral vectors, nanoparticles or physical and mechanical delivery methods like nucleofection, cell squeezing or lipofection that utilize electric field, mechanical force, or cationic lipids for temporary disruption of the cell membrane.119 The latter are primarily suited for therapeutic ex vivo gene editing, while viral
vectors and nanoparticles are mainly used for in vivo gene therapy.12 0
Viral delivery of CRISPR-Cas9 has been explored using lentivirus, adeno-virus, and adeno-associated virus (AAV).105 AAVs are most widely used for gene therapy clinical trials due to their ability to transduce different cells types and tissues, low risk of genomic integration and
low immunogenicity.121 However, AAV limited packaging capacity (-4.5 kb) makes it impossible
to accommodate all the components of CRISPR-spCas9, including sgRNA and a donor DNA template, into a single AAV. Noteworthy is that a host immune response to AAV-CRISPR-Cas9
has been observed in mice, elicited by Cas9 immunogenicity and possibly aggravated by its prolonged expression.12 2
Complementary to the viral systems, an abundance of nanoparticles composed of various bio-compatible materials are being developed for delivery of CRISPR-Cas9. As with their use in protein modulation, nanoparticles for gene editing have demonstrated high loading capacity for nucleic acid cargos, ability to modify payload bio-distribution and pharmacokinetics through an active targeting and formulation, as well as simplicity of manufacturing with a high level of control over their physicochemical parameters e.g. size/shape, kinetics of payload release. 1 23 Nanoparticle-based mRNA delivery of CRISPR-Cas components is therapeutically
attractive due to transient nature of mRNA expression, no risk of genomic integration and mRNA cytoplasmic activity, alleviating the need to overcome nuclear barrier as compared to pDNA (Figure 1-2). To date, nanoparticle-mediated delivery of spCas9 mRNA have been used in combination with AAVs encoding a sgRNA and a repair template to induce repair of Fah gene in a hereditary tyrosinemia in adult animals.9 9 The efficiency of correction was >6% of hepatocytes after a single application, much higher than with a hydrodynamic injection of pDNA (0.4%) previously reported for the same disease.98 Similarly, lung delivery of mRNA encoding zinc finger nucleases (ZFNs) complexed into chitosan-coated nanoparticles, used in combination with AAV6 expressing donor template, resulted in correction of surfactant protein B gene in mice with SP-B deficiency and extended their survival.12 4 Interestingly, the combination
of mRNA nanoparticle with the virus was superior to AAV alone, reaching HDR rates in lung cells ~9%. Recently, a study described the synthesis and development of zwitterionic amino lipids that have been used to formulate nanoparticles capable of simultaneous in vivo delivery of
Cas9 mRNA and sgLoxP to induce expression of floxed tdTomato in the liver, kidneys, and lungs of LSL-TdTomato mice.12 This study shows the potential of nanoparticle-RNA platform to accommodate multiple components of CRISPR-Cas9 into a single carrier, and could possibly be extended to also include a donor template. Lipid and polypeptide nanoparticles have been also used to deliver RNA-protein complex of Cas9 and sgRNAs, which is another promising strategy that ensure transient cellular presence of Cas9 significantly reducing off-target effect.126,127
1.2 Nucleic Acid Delivery using PBAEs
As mentioned in the previous section, polymeric nanoparticles are one of the most widely-used platforms for nucleic acid delivery. PEI, in particular, has been utilized in many studies due to its high cationic charge density and near-neutral pKa amine groups. These chemical features allow it to bind and condense mRNA effectively, as well as promote endosomal buffering and escape. Unfortunately, PEI is not biodegradable, and is therefore subject to concerns regarding long-term use. Moreover, PEI has been shown to be cytotoxic, apparently as a result of the same membrane destabilization that makes it effective in entering cellular cytoplasm.128
Therefore, research into polymeric nucleic acid delivery vehicles is focused on designing polymers with similar properties to PEI (e.g. cationic, high density of amine groups) that are also less cytotoxic (e.g. biodegradable).
In light of the gene-delivery properties of PEI, both positive and negative, Lynn et al. developed poly(#-amino esters), or PBAEs, through conjugate addition of diacrylates and amines in 2000.21 Like PEI, PBAEs are positively charged and have near-neutral pKa amines. Unlike PEI, however, the PBAE backbone is formed of ester bonds that are degradable by esterases within the body, which should further reduce the long-term toxicity of the polymers. This degradation of ester bonds also makes it easier for the nucleic acid payload within the polymers to be unpackaged once the particles reach their destination.
Another advantage of PBAEs is their facile synthesis: one pot polymerization can be performed at relatively benign conditions using commercially available monomers with no purification necessary. The favorable polymer properties and synthetic simplicity of PBAEs have led to a steady increase in their use for biomedical applications (Figure 1-3). Here we review the development of a wide variety of combinatorial material screens which have identified highly potent PBAE materials for a variety of applications, both in vitro and in vivo, including the structure-property relationships they have revealed and the applications towards which they have been developed. We also detail studies which seek to optimize and/or more specifically apply top-performing PBAEs identified in previous screens and novel PBAEs not developed as part of a large library, as well as those which utilize PBAEs for applications other than nucleic acid delivery. 40 30 (620 CL 0. .10 o Z 0 9U 40-I I
Figure 1-3: Number of new PBAE publications each year (data from MEDLINE).
1.2.1 Combinatorial Library Synthesis of PBAEs for Gene Delivery
PBAEs are composed of at least two monomers: a diacrylate and an amine (Figure 1-4). The acrylate groups and nucleophilic amine can undergo a Michael addition without the use of a catalyst in mild conditions,129 and the bifunctionality of the diacrylate monomer allows for a step-polymerization to proceed.130The first reported synthesis of potential biomaterials utilizing step
polymerization via Michael addition between diacrylates and amines was by Danusso and Ferruti in 1970.131 Lynn et al. in 2000 built on this strategy to generate the first PBAEs by reacting 1,4 butanediol diacrylate with the bis(secondary amines) 4, 4'-trimethylenedipiperidine, N,N'-dimethylethylenediamine, and piperazine to synthesize three distinct polymers.2 1 These
polymers were formed in a single step in THF and dichloromethane at 500C, and were subjected to simple purifications in diethyl ether and/or hexane with yields of up to 86%. Importantly, all 3 polymers were capable of binding with DNA plasmids and condensing them into nanometer-scale particles, though only one of the polymers was capable of effective in vitro gene transfection relative to PEI.
a
b
0 O 0
IR
R
R'- NH2
C 0DNA mid PBAE
O ONH2 NH 0 H NH * NH 0 SN" - NH2 O NH HO M0 N H NH 2 0 0 0 0 NH2 NH2 -_Ol C: 001_ H 0 N * NH2 0O0- H- o -H HN j 0 N
Figure 1-4. PBAE and nanoparticle synthesis. (a) Representative PBAE synthesis. Diacrylate and amine monomers step-polymerize via a Michael addition. (b) PBAEs bind and condense DNA plasmids (and other nucleic acids) into nanoparticles via charge interactions between cationic amine groups and the anionic nucleic acid. (c) A set of monomers used in one of the first combinatorial library screens of PBAE materials, adapted from Lynn et al.1 32
Based on the promise of the original PBAEs, along with the evidence that monomer identity plays an important role in polymer efficacy, Lynn and colleagues sought to improve upon the PBAE platform for gene delivery. The specific biological mechanisms by which nanoparticles
deliver nucleic acids and the subtle structural cues that enable them has yet to be elucidated, 10,133,134 making rational design, especially based off of such a small set of initial
polymers, a challenge. As a result, parallel screening approaches of chemically diverse PBAEs have been adopted to improve the potency of gene delivery while potentially elucidating important structure-function relationships. PBAEs lend themselves incredibly well to library development using combinatorial synthesis due to the following factors:135
1) A wide chemical diversity of diamine and diacrylate monomers are commercially available
2) Polymerization requires only a single step
3) Purification is unnecessary as no byproducts are generated
With these factors in mind, Lynn et al. developed a modest library of 140 PBAEs synthesized from 7 diacrylate and 20 bis(secondary) or tertiary amine monomers.135 Following
nanoprecipitation with DNA and transfection of COS-7 cells, multiple materials were demonstrated to outperform the commercially available transfection reagents lipofectamine and PEI. Altogether, the polymer synthesis and initial screen took approximately two weeks.
The success of this initial screen, both in terms of material potency and scalability of library design, set the stage for over a decade of research into the development of PBAE libraries, and has even influenced the development of libraries for other nucleic acid delivery materials, notably lipid nanoparticles for siRNA delivery.31
,33,35 Indeed, Anderson et al. showed that library synthesis could be further scaled up, synthesizing over 2500 polymers in a semi-automated fashion using commercially available diacrylates and amines with various structures and functional groups." From this screen, 33 polymers were identified that outperformed PEI for DNA delivery to COS-7 cells. Many of these successful polymers formed the basis for libraries tailored for more specific applications, which are discussed in later sections.
Several other classes of PBAE materials have also been developed using the library screening approach. One such example is the hyperbranched PBAE. As opposed to the traditional linear PBAEs, hyperbranched PBAEs (hPAEs) can be synthesized using trifunctional monomers to facilitate branching, which allows for greater synthetic flexibility that may lead to key advantages in nucleic acid delivery.1 36 Zhong et al. developed a small library of hPAEs
using the trifunctional amines 4-(aminomethyl)piperidine, 1-(2-aminoethyl)piperazine, and N-methylethylenediamine which demonstrated comparable potency and superior toxicity compared to PEI, although the study made no direct comparisons to linear PBAEs. The synthetic potential of hPAEs was greatly expanded through the development of the so-called "A2+B3/B2" strategy (Figure 1-5a) by Cutlar et al. which utilizes trimethylolpropane triacylate as a branching agent, allowing any diacrylate/amine combinations from previous screens to be branched.1 37 Polymers prepared via this method were shown to have enhanced colloidal
stability and DNA transfection compared to top-performing linear PBAEs.
Other material libraries have included variations on the linear approach. Our lab has reported the synthesis of PBAE terpolymers (Figure 1-5b), which incorporate an amine-terminated hydrocarbon chain in addition to the traditional diacrylate and amine.138 These
materials are not only capable of non-covalent formulation with hydrophobic moieties, analogous to leading lipid nanoparticles,' 37 but were shown to be inherently more potent than previous generations of linear PBAEs.
Deng et al. developed a small library of PBAEs with light-sensitive 2-nitrobenzene diacrylates (Figure 1-5c) that degraded on a scale of minutes when exposed to UV irradiation.1 39
Incorporation of bioreducible monomers, such as cystamine which can trigger release of cargo in intracellular reducing environment (Figure 1-5d), was also investigated in screens by Tzeng et
al.2 3
shown modest increases in transfection capability over traditional materials for both gene delivery"' and siRNA delivery."
a
'+
b
c
d
PIC C
Figure 1-5. Alternate PBAE Syntheses. (a) Hyperbranched PBAEs synthesized using the "A2+B3/B2" method, adapted from Cutlar et al.137 (b) PBAE terpolymer synthesis, which utilizes an amine-terminated
alkane chain within the polymer, adapted from Eltoukhy et al.138 (c) Photo-sensitive PBAEs synthesized
with 2-nitroenzne diacrylates, adapted from Deng et al.1 39 (d) Cystamine-terminated PBAEs, adapted
from Tzeng et al.23
The ability to quickly and economically identify lead compounds for nucleic acid delivery is only a part of the utility of PBAE screens. The other advantage to screening lies in the ability to rapidly assess, and ultimately optimize, PBAE synthesis based on the identification of structure-function relationships. That is, PBAE libraries can be iteratively optimized based on synthetic trends observed in previous libraries. Broadly speaking, the synthetic handles for PBAEs most commonly assessed are monomer chemistry, polymer molecular weight, and polymer end-capping.
As a follow-up to their large screen of over 2000 PBAEs, and as an expansion on a previous study which investigated the effect of these synthetic handles,1
12 Anderson et aL
synthesized a smaller screen (486 polymers) using a subset of the most potent diacrylates and amines identified in the large library.143The study demonstrated that, in general, hydrophobic
diacrylates were the most potent, which a later screen by Sunshine et al. of >300 similar polymers has corroborated.144 This trend may be explained in part by an increase in beneficial
hydrophobic interactions with the cell and endosomal membrane,145
,141 which could in turn help
explain why terpolymers, which incorporate long-chain hydrophobic monomers, are more potent than many "standard" PBAEs.1 38 Studies with hPAEs have also benefitted from the use of
hydrophobic diacrylates,137147 although a direct comparison with more hydrophilic monomers
has yet to be made for these materials.
The Anderson et al. library not only sought to elucidate relationships between monomer identity and efficacy, but also included various amine:acrylate monomer ratios as a means of altering the polymer molecular weight.130 The closer the monomer ratio is to unity, the higher the
molecular weight of the polymer, although precise control over molecular weight for step-growth polymers is typically difficult to achieve.130 Still, if polymers are made at a large enough scale for
more accurate control over monomer ratio to be possible, researchers can (and have) made some important observations regarding the relationship between PBAE molecular weight and efficacy. The Anderson et al. functional screen found no polymers possessing high transfection capabilities below a size of 10 kDa,143 which agrees with results that demonstrated a positive
correlation between PEI molecular weight and its efficacy as a nucleic acid delivery vehicle.14 8
The importance of molecular weight is dependent on the type of material, as evidenced by the lack of a precisely linear trend in the aforementioned screen, though for some PBAEs even a slight change in monomer ratio can result in order of magnitude changes in nanoparticle efficacy.149 Interestingly, our lab has shown that, for certain crude PBAEs there exists an
intermediate "optimum" molecular weight in terms of transfection efficiency but when the crude polymer is fractioned based on molecular weight, efficacy monotonically increases with molecular weight." 0 For linear PBAEs, the increases in efficacy with molecular weight often
come at the cost of increased cytotoxicity, but a recent study has demonstrated that higher molecular weight may increase cellular viability for some hPAEs.14 7
In addition to affecting the molecular weight of the polymer, monomer ratio also determines the unit that terminates the polymer chain: In a two monomer system, the monomer that is in excess will represent the vast majority of the polymer end caps.1 30 Thus, in varying the
monomer ratio, Anderson et al. were also able to observe that amine-terminated polymers (i.e. polymers with a diacrylate:amine ratio less than one) far outperformed their acrylate-terminated counterparts.14 3 This phenomenon may be caused by a reduced cellular uptake of
acrylate-terminated polymers.151 Furthermore, the identity of the end-capping amine can be made
different than that of the backbone amine if the polymerization proceeds in two steps: i) an initial polymerization with an excess of diacrylate monomer, followed by ii) reaction of the acrylate-terminated polymer with an excess of end-capping amine. An initial study demonstrated that the identity of the end-capping amine has order of magnitude consequences on ultimate PBAE efficacy, and that the optimal end-capping amines were not the same as those that made up the most effective polymer backbone.152 Multiple subsequent libraries have confirmed this
effect,23
,144,153,154
and even subtle changes to the end-capping chemical identity can have important consequences for PBAE efficacy.155 hPAE efficacy has also proven to be dependent
on terminal amine groups,156 a dependence which may be amplified by the increased density of
end groups that result from the hyperbranched structure. It is worth noting that, in light of this data, the vast majority of recent and current PBAE projects utilize the two-step synthesis in order to control the end-capping amine.
Although most structure-function screens tend to focus on how polymer structure affects its nucleic acid delivery capacity, there are some screens that have focused specifically on other aspects of PBAE performance. One such property is polymer degradation, which has implications for both biological clearance and endosomal escape. As previously mentioned, a