Transport / adsorption / diffusion / agrégation d'atomes pulvérisés par plasma dans les matériaux poreux. Expériences et modélisations
18
0
0
Texte intégral
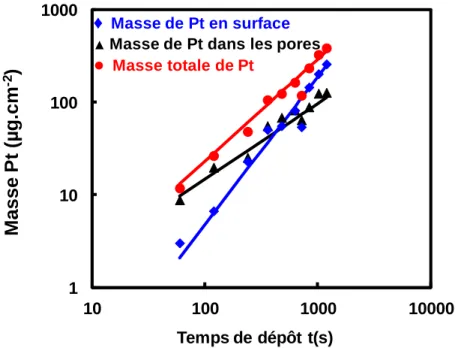
Figure




+7


Documents relatifs