Chiral-at-Metal BODIPY-Based Iridium(III) Complexes: Synthesis and Luminescence Properties
Texte intégral
Figure

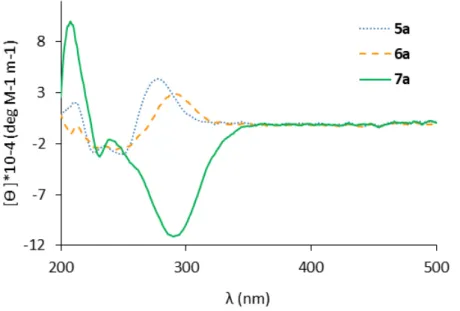



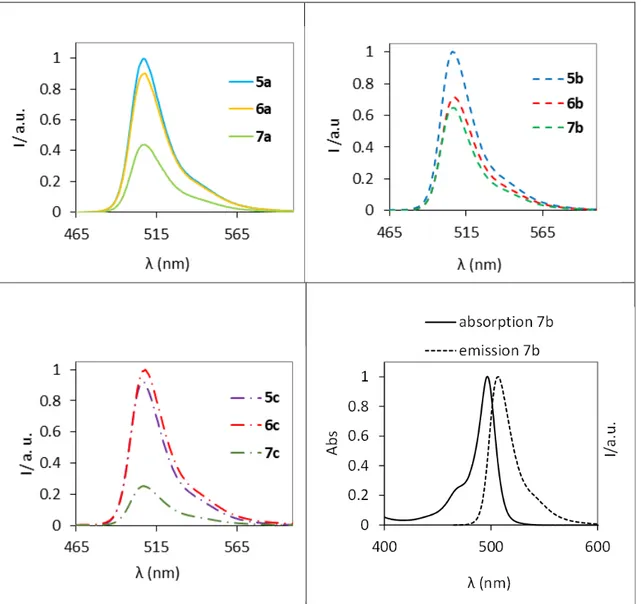

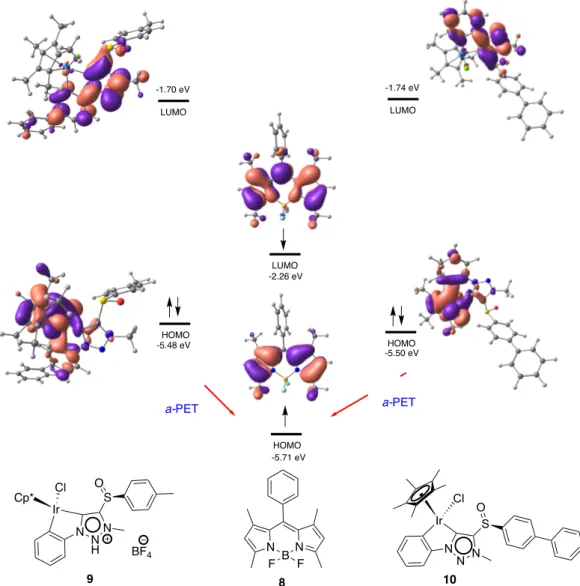
Documents relatifs
Chen, Gao and coworkers designed a NP-based trimodal probe including dipicolylamine-Mn(II) complex as MRI probe and anticancer agent targeting HIF and LDH and BODIPY
8 Herein, we describe an efficient strategy for the synthesis of robust chiral luminescent octahedral NHC-iridium(III) complexes and enantio-enriched
The resulting reaction mixture was stirred at rt for 30 min, then filtered through celite and concentrated under reduced pressure.. The resulting reaction mixture
In addition, in terms of deictic perspective too, the persistence is hypothesized to be stronger in DIST: while neither FIST nor DIST involves the full deictic perspective shift
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des
The fraction of residual strain hardening with annealing time for the isothermal annealing is shown in Fig. The experimental data clearly show that the addition of Sn solute reduces
The presence of oxyallyl patterns in such ligands is expected to confer the redox-activity and can favor the ligand centered chemical oxidations/reductions in the formed
The previous section summarizes the research done in the past twenty years on tridimensional Fe/Co Prussian Blue Analogues, which display thermally and
