A structural study of copper(II) carboxylates: Crystal structure and physical characterisation of [Cu2(2-bromopropanoato)4(caffeine)2]
10
0
0
Texte intégral
Figure
![Table 1. Summary of Crystal Data, Intensity Measurement and Structure Refinement for [Cu 2 (2-bromopropionato) 4 (caffeine) 2 ]](https://thumb-eu.123doks.com/thumbv2/123doknet/14868490.638615/3.891.449.770.152.332/summary-crystal-intensity-measurement-structure-refinement-bromopropionato-caffeine.webp)
![Fig. 1. Ortep view of the crystal structure of [Cu 2 (2- (2-bromopropionato) 4 (caffeine) 2 ] showing the atom number-ing of the asymmetric unit (black bonds) and the part of the binuclear complex generated by a centre of inversion (white bonds)](https://thumb-eu.123doks.com/thumbv2/123doknet/14868490.638615/4.891.478.798.118.527/crystal-structure-bromopropionato-caffeine-asymmetric-binuclear-generated-inversion.webp)
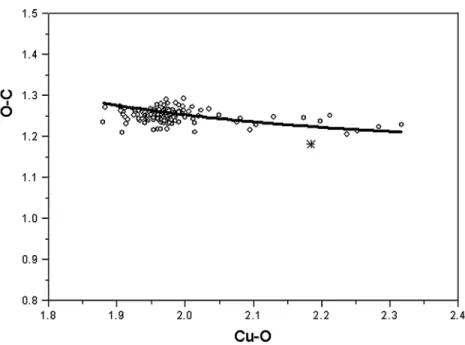
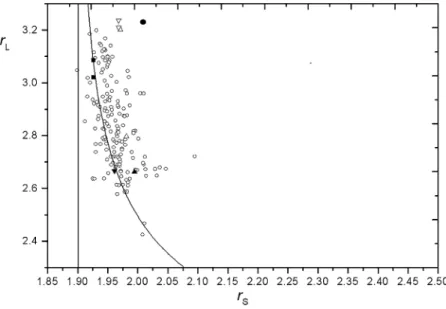
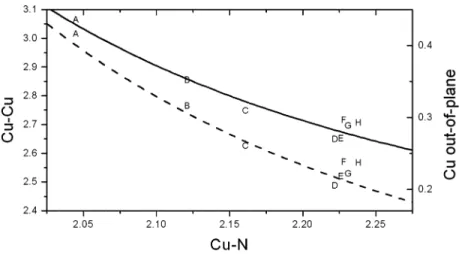
+3
![Table 3. Selected structural parameters of [Cu 2 X 4 (caffeine) 2 ] complexes](https://thumb-eu.123doks.com/thumbv2/123doknet/14868490.638615/8.891.122.803.142.311/table-selected-structural-parameters-of-cu-caffeine-complexes.webp)
Documents relatifs