HAL Id: tel-02865552
https://tel.archives-ouvertes.fr/tel-02865552
Submitted on 11 Jun 2020HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Jules Russick
To cite this version:
Jules Russick. Traitement de l’hémophilie A à l’aide d’ARNm codant le facteur VIII et prévention de la réponse immunitaire dirigée contre le facteur VIII thérapeutique. Immunologie. Sorbonne Université, 2018. Français. �NNT : 2018SORUS362�. �tel-02865552�
THESE DE DOCTORAT DE L’UNIVERSITE
SORBONNE UNIVERSITE
Ecole Doctorale : Physiologie, Physiopathologie et Thérapeutique (ED394) Spécialité : Immunologie
Présentée par :
Jules RUSSICK
Pour l’obtention du grade de
Docteur de l’université Sorbonne Université
Sujet de la thèse
Traitement de l’hémophilie A à l’aide d’ARNm codant le FVIII
et prévention de la réponse immunitaire anti-FVIII
Soutenue le 17 Septembre 2018
Devant le jury composé de :
Mme Isabelle CREMER, Professeur, Université Sorbonne Université (Présidente)
M Bernard MAILLERE, Directeur de recherche, CEA (Rapporteur)
L’hémophilie A est une maladie rare liée au chromosome X due à l’absence de facteur VIII (FVIII) fonctionnel de la coagulation. Elle est causée par une mutation du gène F8 et se traduit par des symptômes cliniques plus ou moins invalidants selon la sévérité de la maladie. On constate l’apparition d’hémorragies ponctuelles suite à un traumatisme pour les formes les plus légères, des hémorragies spontanées au niveau des articulations (hémarthroses) ou des tissus mous (hématomes) qui peuvent provoquer des arthropathies invalidantes ou mettre le pronostic vital en jeu. La prévention ou le traitement des saignements se fait par injection de FVIII thérapeutique recombinant. L’injection de ce FVIII exogène provoque, chez 25 à 30% des patients, l’apparition d’une réponse immunitaire anti-FVIII. Cette réponse immunitaire se traduit par l’apparition d’anticorps anti-FVIII qui neutralisent l’activité pro-coagulante du FVIII, appelés donc « inhibiteurs ». Cette complication de la thérapie de substitution mène les patients à une impasse thérapeutique. La réponse anti-FVIII est décrite comme une réponse immunitaire T-dépendante classique au cours de laquelle le FVIII est endocyté et apprêté par des cellules présentatrices d’antigène. Les peptides du FVIII sont présentés sur le complexe majeur d’histocompatibilité de classe II (CMH II) et reconnus par des lymphocytes T naïfs via leur récepteur (TCR). Ils s’activent et activent à leur tour des lymphocytes B via leur récepteur (BCR) qui, après activation, se différencient en plasmocytes et synthétisent les inhibiteurs, ou en lymphocytes B mémoires qui, lors d’une nouvelle rencontre du FVIII, pourront déclencher une réponse plus forte et plus rapide. La stratégie la plus couramment utilisée pour traiter les patients avec inhibiteurs l’induction de tolérance immunitaire (ITI) et consiste en l’injection répétée de fortes doses de FVIII sur une longue période. Cependant, ce traitement est extrêmement contraignant pour le patient, est très coûteux et n’est efficace que dans 60 à 80% des cas. Au cours de ma thèse, j’ai tout d’abord souhaité proposer et valider une nouvelle stratégie thérapeutique, alternative à la thérapie de substitution, utilisant un ARNm codant le FVIII. La courte demi-vie du FVIII (environ 12h) oblige les patients à des injections régulières de FVIII. L’utilisation d’un ARNm codant le FVIII permettrait une production plus longue de FVIII, sans se confronter aux difficultés induites par la thérapie génique. J’ai ainsi montré que les ARNm codant le FVIII, lorsqu’ils sont formulés dans des nanoparticules lipidiques (LNP), permettent la production de FVIII endogène in vitro et in vivo. De plus,
production hépatique. Enfin, j’ai démontré qu’une seule injection d’ARNm permet une correction totale du phénotype hémorragique chez la souris déficiente en FVIII.
Le deuxième aspect de ma thèse a consisté à étudier la possibilité d’inhiber la réponse immunitaire anti-FVIII par inhibition des lymphocytes B via l’inhibition de la voie de signalisation du BCR. Avec Sandrine Delignat, nous avons ainsi étudié l’effet d’un inhibiteur de la Bruton’s Tyrosine Kinase (Btk), une kinase centrale dans l’activation du lymphocyte B. Nous avons ainsi montré que l’inhibition de la Btk ne permettait pas de prévenir l’apparition d’une réponse immunitaire dirigée contre le FVIII. En revanche, des expériences de transfert adoptif chez des souris déficientes en FVIII nous ont permis de montrer que cette inhibition permet d’altérer la réponse mémoire anti-FVIII. En effet, les souris qui ont reçu des lymphocytes B mémoires spécifiques du FVIII ne développent pas de réponse mémoire lorsqu’elles sont traitées avec l’inhibiteur de la Btk, en plus du FVIII. A l’inverse, on constate l’apparition d’inhibiteurs chez les souris simplement traitées avec du FVIII après le transfert de lymphocytes B mémoire. Des expériences in vitro nous ont également permis de montrer que cette inhibition de la Btk avait pour effet l’inhibition de la différenciation des cellules B mémoires en plasmocytes.
Enfin, la dernière partie de ma thèse m’a permis de caractériser une lignée de souris hémophiles transgéniques pour l’allèle HLA-DRB1*01:01 du CMH II humain. J’ai en effet caractérisé des souris déficientes en FVIII, pour les CMH I et II murins, mais exprimant les CMH I et II humains. J’ai ainsi montré que ces souris naïves produisaient plus d’IgM et moins d’IgG que les souris hémophilies exprimant les CMH murins. J’ai également montré que cela est associé à une modification du nombre de lymphocytes B folliculaires, mais pas du nombre de cellules présentatrices d’antigène. De plus, les lymphocytes T ont la même capacité à proliférer dans les deux lignées. J’ai ensuite caractérisé la réponse immunitaire anti-FVIII chez ces souris et j’ai confirmé la prévalence des IgM anti-FVIII par rapport aux IgG anti-FVIII et confirmé la même prolifération des lymphocytes T en réponse au FVIII. J’ai étudié l’expression du CMH humain par les cellules présentatrices d’antigène de ces souris et, si j’ai confirmé la perte d’expression du CMH murin, je n’ai pas détecté d’expression du CMH humain.
mRNA) permet la production de molécules endogènes, même dans le cas où, comme pour le FVIII, les taux de traduction sont très faibles. Des études restent cependant à mener quant à l’immunogénicité de cette stratégie, ainsi qu’à la possibilité d’améliorer le ciblage des cellules transféctées après injection. De même, des études approfondies sont à mener pour améliorer la stratégie d’inhibition de la Btk. Si ce traitement permet l’inhibition de la réponse mémoire, il serait intéressant d’améliorer sa spécificité. L’inhibition ne touche que les lymphocytes B qui fixent un antigène sur le BCR, mais quel que soit cet antigène. Enfin, la possibilité d’avoir un modèle murin exprimant un CMH II humain ouvre la voie à de très nombreux projets. En effet, une étude des peptides présentés par le HLA-DRB1*01:01 deviendrait possible, ainsi que la génération de FVIII muté sur les peptides T reconnus par cet allèle. Des études complémentaires sont néanmoins nécessaires afin de comprendre les mécanismes responsables de l’absence de CMH II à la surface des cellules de cette lignée transgénique.
Ce travail de thèse repose sur les publications suivantes :
Article 1 : Jules Russick, Sandrine Delignat, Peter Milanov, Olivier Christophe, Gabor Boros, Katalin Kariko, Srini V Kaveri, Sébastien Lacroix-Demazes
Correction of bleeding in experimental severe hemophilia A by systemic delivery of factor VIII-encoding mRNA (Soumis à Blood et refusé, en cours de réécriture)
Article 2 : Sandrine Delignat, Jules Russick, Bagirath Gangadharan, Julie Rayes, Mathieu Ing, Jan Voorberg, Srinivas V Kaveri, and Sébastien Lacroix-Desmazes
Prevention of the anti-factor VIII memory B-cell response by inhibition of the Bruton’s tyrosine kinase in experimental hemophilia A (Soumis à Haematologica)
Article 3 : Jules Russick, Sandrine Delignat, Yu-Chun Lone, Marc Pallardy, Srinivas V Kaveri, Sébastien Lacroix-Desmazes
Generation of humanized FVIII-deficient HLA-DRB1*01:01-transgenic mice (En préparation, non soumis)
INTRODUCTION ... 13 I. HÉMOPHILIE A ET FVIII ... 14 1.HISTORIQUE DE L’HEMOPHILIE --- 14 2.DESCRIPTION DE L’HEMOPHILIE A --- 14 3.GENETIQUE DE L’HEMOPHILIE A --- 15 Le gène F8 ... 15
Anomalies génétiques menant à l’hémophilie A ... 16
4.CYCLE DE VIE DU FVIII --- 17
Description du FVIII ... 17
Synthèse du FVIII ... 18
Demi-vie du FVIII ... 19
Rôle du FVIII ... 20
Catabolisme du FVIII... 23
II. TRAITEMENTS DE L’HEMOPHILIE A ET OPTIONS THERAPEUTIQUES ... 24
1.TRAITEMENTS ACTUELS --- 24
FVIII thérapeutiques ... 24
Thérapies sans FVIII ... 25
2.NOUVELLES STRATEGIES --- 26
FVIII à longue durée de vie ... 27
Emicizumab ... 29
Thérapie génique... 31
III. COMPLICATION DU TRAITEMENT ... 33
1.IMMUNOGENICITE DU FVIII THERAPEUTIQUE --- 33
2.FACTEURS DE RISQUES INTRINSEQUES AUX PATIENTS --- 34
Anomalies du gène codant le FVIII ... 34
Autres gènes ... 35
Ethnie ... 35
3.AUTRES FACTEURS DE RISQUES --- 36
Antécédent de traitement ... 36
Le FVIII lui-même ... 36
Intensité du traitement ... 37
Traitement prophylactique ou à la demande ... 38
Changement de produit ... 40
4.REPONSE IMMUNITAIRE --- 42
Description générale de la réponse anti-FVIII thérapeutique ... 45
Endocytose et apprêtement du FVIII ... 46
CD206 et récepteurs d’endocytose ... 47
vWF et endocytose ... 48
FVIII à longue durée de vie et endocytose ... 50
Présentation aux lymphocytes T ... 51
Rôle des lymphocytes B ... 54
IV. STRATEGIES D’INDUCTION DE TOLERANCE AU FVIII ... 57
1.INDUCTION DE TOLERANCE IMMUNITAIRE (ITI) --- 57
Description des protocoles d’ITI ... 57
II. PREVENTION DE LA REPONSE MEMOIRE ANTI-FVIII PAR INHIBITION DE LA ‘BRUTON’S
TYROSINE KINASE’ (BTK) CHEZ LA SOURIS DEFICIENTE EN FVIII ... 102
III. CARACTERISATION D’UN NOUVEAU MODELE MURIN DEFICIENT EN FVIII ET HUMANISE POUR LE HLA-DRB1*01:01... 132
DISCUSSION ... 153
CONCLUSION & PERSPECTIVES ... 171
Figure 1. Production du FVIII. 16
Figure 2. Clivage intracellulaire du FVIII. 19
Figure 3. Activation du FVIII. 21
Figure 4. Cascade de la coagulation. 22
Figure 5. Régulateurs naturels de la cascade de la coagulation
et stratégies pour les inhiber. 26
Figure 6. FVIII à longue durée de vie et essais cliniques correspondants. 29
Figure 7. Schéma du mode d’action du FVIIIa ou de l’Emicizumab. 30
Figure 8. Représentation schématique de la théorie du danger. 38
Figure 9. Représentation schématique de la théorie
de la discontinuité de l’antigène. 39
Figure 10. Facteurs de risques liés à l’apparition d’inhibiteurs. 41
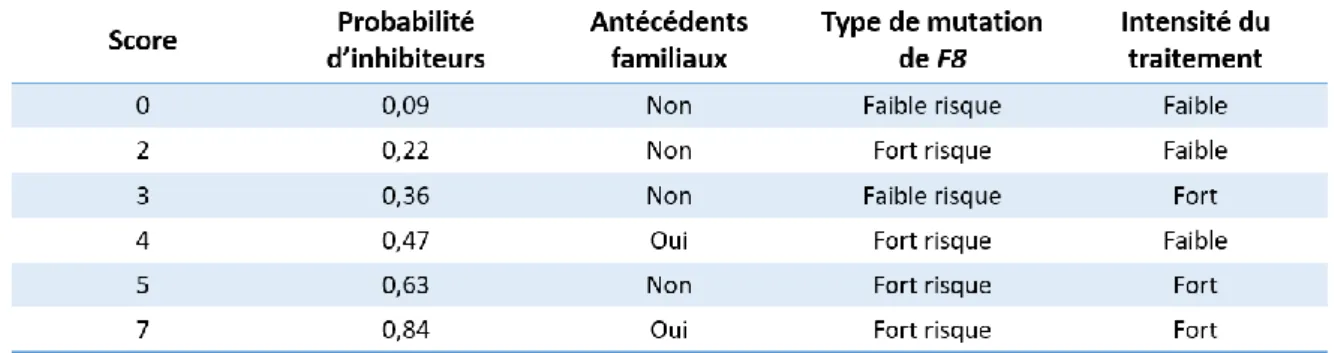
Figure 11. Score clinique CANAL (Concerted Action on Neutralizing
Antibodies in patients with severe haemophilia A). 42
Figure 12. Réponse immunitaire anti-FVIII. 46
Figure 13. Récepteurs capables de fixer le FVIII et/ou le vWF 50
aa acide aminé
AAV Adeno-Associated Virus ABR Annual Bleeding Rate
ADCC Antibody-dependent cell-mediated cytotoxicity AID Activation-Induced cytidine Deaminase
aPC Activated Protein C
aPCC activated Prothrombin Complex Concentrates ARN Acide Ribonucléique
ARNm ARN messager ARNt ARN de transfert AT Antithrombine BCR B Cell Receptor
BDD FVIII B Domain Deleted FVIII Btk Bruton’s Tyrosine Kinase
CANAL Concerted Action on Neutralizing Antibodies in patients with severe haemophilia A
CDC Complement-Dependent Cytotoxicity CG Centre Germinatif
CHAMP CDC Hemophilia A Mutation Project CHO Chinese Hamster Ovary
CMH (I ou II) Complexe Majeur d’Histocompatibilité (de classe I ou II) CPA Cellule Présentatrice d’Antigène
CTL Cytotoxic T Lymphocyte
CTLA-4 Cytotoxic T Lymphocyte Antigen 4 DC Dendritic cells
FT Facteur Tissulaire FVIII(a) Facteur VIII (activé) HIGM Syndrome d’Hyper IgM HLA Human Leukocyte Antigen IFNγ Interféron gamma
Ig (G) Immunoglobuline (de classe G) IgIV Immunoglobulines Intraveineuses IL Interleukines
ITAM Immunoreceptor Tyrosine-based Activation Motif ITI Induction de Tolérance Immunitaire
iTreg induced Treg
IVT mRNA In vitro Transcribed mRNA
LB Lymphocyte B LNP Lipid NanoParticle LT (h) Lymphocytes T (helper) miRNA micro RNA
MO-DC Monocyte-derived Dendritic Cells NK cells Natural Killer cells
nTreg natural Treg
PBMC Peripheral Blood Mononuclear Cells pdFVIII Plasma Derived FVIII
PLCγ2 Phospholipase C-γ2
PTPs Previously Treated Patients PUPs Previously Untreated Patients
siRNA short interfering mRNA TCR T Cell Receptor
Teff LT effecteur
Tfh LT follicular helper
TFPI Tissue Factor Pathway Inhibitor TLR Toll-Like Receptor
TNFα Tumor Necrosis Factor α Treg LT régulateur
UB Unité Bethesda
UPR Unfolded Protein Response VHC Virus de l’Hépatite C
VIH Virus de l’Immunodéficience Humaine vWF Facteur Willebrand
I. Hémophilie A et FVIII
1. Historique de l’hémophilieBien avant sa description et sa caractérisation, l’hémophilie est évoquée dès le Ier siècle
de notre ère. Rabban Shimon ben Gam liel (environ -10 av. JC – 70 ap. JC) interdit en effet la circoncision d’un garçon, car les fils de trois de ses tantes sont décédés de saignements consécutifs à l’acte. Un siècle plus tard, Rabbi Juda Ha-Nassi autorise dans la Mishna (première partie du Talmud), qu’un bébé de sexe masculin ne soit pas circoncis, si deux de ses frères sont déjà décédés de saignements suite à cette pratique1,2.
Au cours du XIXe siècle, de nombreuses appellations sont utilisées pour décrire ce
syndrome : idiosyncrasie hémorragique, hématophilie, diathèse hémorragique héréditaire, maladie hémorragique… Pour la première fois, Hopff utilise en 1828, le terme d’ « hémophilie » - haima (sang en grec) et philia (amour)3. C’est également au cours de
ce siècle que nait la Reine Victoria du Royaume-Uni, porteuse asymptotique d’une mutation causant l’hémophilie. Sa descendance étant présente dans la majorité des cours d’Europe, on trouvera cette mutation dans de très nombreuses familles royales, jusqu’à celle du dernier tsar de Russie. C’est ce qui vaudra à l’hémophilie d’être appelée « la maladie Royale ». Cependant, c’est de l’hémophilie B dont il s’agit ici comme cela a récemment été découvert4; depuis 1952 et les travaux d’Aggeler5 et de Biggs6, deux types
d’hémophilie sont connus : l’hémophilie A si le gène codant le facteur VIII (FVIII) est invalidé et hémophilie B si c’est celui codant le facteur IX (FIX).
2. Description de l’hémophilie A
L’hémophilie A est une maladie hémorragique qui affecte environ 1 naissance masculine sur 5 0007, ce qui en fait une des maladies hémorragiques les plus fréquentes8. Elle est
causée par différentes anomalies du gène F8 codant pour le FVIII de la coagulation. Ces mutations induisent un déficit quantitatif et/ou qualitatif de FVIII, ce qui se traduit par une importante hétérogénéité phénotypique de la maladie. Selon l’activité résiduelle du FVIII, l’hémophilie est qualifiée de sévère si l’activité résiduelle est inférieure à 1% de celle d’un plasma normal, de modérée entre 1 et 5% ou de mineure si elle est supérieure à 5%9.
Ces sévérités concernent respectivement 50%, 10% et 40% des patients hémophiles A10
et se traduisent par des manifestations cliniques très diverses. Dans les cas les plus légers, les hémorragies sont ponctuelles et font souvent suite à un traumatisme ; par exemple lors d’une petite chirurgie. En revanche, dans les formes les plus sévères de la maladie, des hémorragies peuvent advenir spontanément et de manière chronique. Des hémorragies internes apparaissent en particulier au niveau des articulations (hémarthroses) pouvant déclencher des arthropathies invalidantes et conduire à la paralysie. Ces hémarthroses, très caractéristiques de l’hémophilie, ont d’ailleurs été décrites dès 1890 par König11.
Des hémorragies se développent également au niveau des tissus mous (hématomes) qui, lorsqu’elles concernent le cerveau, peuvent engendrer des séquelles neurologiques voire mettre en jeu le pronostic vital12. En effet, les hémorragies intracérébrales concernent
7,5% des hémophiles13 et sont 40 à 80 fois plus fréquentes chez les nouveau-nés
hémophiles que dans la population générale14.
3. Génétique de l’hémophilie A
L’hémophilie A est une maladie qui touche presque exclusivement les hommes. C’est ce qu’observe Christian Friedrich Nasse en 1820 en étudiant des familles sujettes à des saignements mortels. Il constate également que les hommes, bien qu’ayant une « disposition héréditaire aux saignements mortels », ne transmettent pas la maladie. C’est la première description de la transmission récessive de l’hémophilie A.
Le gène F8
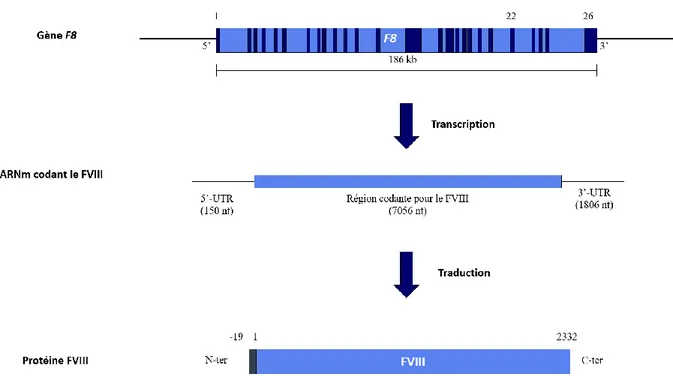
Nous savons aujourd’hui que le gène F8 codant pour le FVIII est situé en position 28 du bras long du chromosome X (Xq28) et que son expression est récessive ; ce qui explique qu’en cas de mutation délétère sur un chromosome X, les femmes soient porteuses de la maladie tandis que les hommes en sont atteints. Ce propos est cependant à nuancer par de rares exceptions de femmes atteintes d’hémophilie A, notamment à cause d’un défaut d’inactivation du chromosome X portant la version muté de F815. Le gène codant le FVIII
traduite (untranslated – 5’ UTR) de 150 nucléotides et d’une région 3’ UTR de 1806 nucléotides, encadrant la région codante de 7056 nucléotides. Cette région codante est ensuite traduite en une protéine de 2332 acides aminés (aa) précédée d’une séquence signal de 19 aa permettant l’adressage de la protéine au réticulum endoplasmique (RE) puis sa sécrétion.
Figure 1. Production du FVIII. Chez l’homme, le gène F8 (bleu clair) est situé en position Xq28 du chromosome X, fait 186 kb et est composé de 26 exons (bleu foncé). Sa transcription engendre un ARNm de 9012 nucléotides (nt) dont 7056 constituent la région codante. La traduction de cette région codante donne une chaine polypeptidique de 2351 acides aminés (aa) dont les 19 premiers constituent un peptide signal d’adressage au réticulum endoplasmique (gris foncé).
Anomalies génétiques menant à l’hémophilie A
A l’heure actuelle, des bases de données telles que « CDC Hemophilia A Mutation Project (CHAMP) » listent près de 3 000 mutations du gène F8 et y associent la sévérité de l’hémophilie observée17.
La grande majorité des mutations sont des mutations ponctuelles, c’est-à-dire qu’elles concernent un seul nucléotide. Elles ont des conséquences variables et résultent en une sévérité de la maladie différente. Les mutations non-sens (11,1% des mutations dans CHAMP), qui engendrent l’apparition d’un codon-stop et aboutissent donc à une protéine tronquée, sont associées dans 90% des cas à une hémophilie A sévère. En revanche, les
mutations faux-sens (49%), qui modifient l’acide aminé synthétisé, résultent-elles en des phénotypes différents. Cette variabilité dépend de la région de la protéine concernée et des différences physico-chimiques entre l’acide aminé normal et celui qui le remplace (hydrophobicité, encombrement stérique, polarité). En effet 38% des patients avec ce type de mutation développent une hémophilie A mineure, soit presque autant que de patients chez qui cela engendre une hémophilie A sévère (39%).
Les délétions sont le deuxième type de mutations les plus fréquentes chez les hémophiles A. Elles concernent de petits (<50pb) ou de larges (>50pb) fragments d’ADN, sont donc associées à des sévérités différentes et représentent 23% des mutations de la base de données CHAMP. Ce pourcentage est plus important dans CHAMP que ce que l’on trouve dans la littérature où elles sont estimées entre 5% et 10% 18.
Concernant les hémophilies A sévères spécifiquement, elles sont consécutives dans 45% des cas à un réarrangement génique qui est l’inversion de l’intron 2219–22. Cette inversion,
qui est due à la recombinaison de deux régions homologues, mène à la transcription des exons 1 à 22 dans le sens 3’-5’ et des exons 22 à 26 dans le sens 5’-3’, ce qui aboutit à la synthèse de deux ARNm incomplets.
Enfin, d’autres mutations peuvent modifier le cadre de lecture, ajouter/éliminer des sites de glycosylation de la protéine ou modifier des sites d’épissage. A noter également que dans certains cas, l’hémophilie A n’est pas causée par une mutation dans le gène F8 à proprement parlé, mais dans ses séquences régulatrices, voire dans d’autres gènes. En effet, certaines mutations concernent des gènes codant des protéines intervenant dans le cycle de vie du FVIII, comme LMAN-1/ERGIC-5323 ou le facteur Willebrand (vWF)24.
4. Cycle de vie du FVIII
Description du FVIII
Le FVIII est un cofacteur de la coagulation principalement synthétisé dans le foie25–27. Ces
résultats ont été confirmés et affinés plus récemment, où des études utilisant soit la détection de l’ARNm codant le FVIII, soit une invalidation conditionnelle de F8, ont démontré une production de FVIII par les cellules endothéliales sinusoïdales28. Il a
extra-de détecter une production extra-de FVIII par cet organe31. De manière générale, le FVIII semble
produit par les cellules endothéliales microvasculaires, principalement du foie, mais également de certains autres organes32.
Le FVIII est une glycoprotéine traduite sous forme d’une chaine polypeptidique de 2351 aa. Elle est alors composée d’un peptide signal de 19 aa à son extrémité N-terminale suivi d’une séquence de 2332 aa divisée en 6 domaines (A1-A2-B-A3-C1-C2). Les trois premiers domaines sont séparés par 30 à 40 aa riches en acides aspartiques (Asp) et acides glutamiques (Glu), appelées « régions acides »33 et nommées a1, a2 et a3. Les domaines A
possèdent environ 40% d’homologie entre eux, avec le facteur V de la coagulation et avec la céruloplasmine34. De la même façon, les domaines C présentent des homologies avec
les domaines C du facteur V. Le domaine B lui, ne montre aucune homologie avec d’autres protéines35.
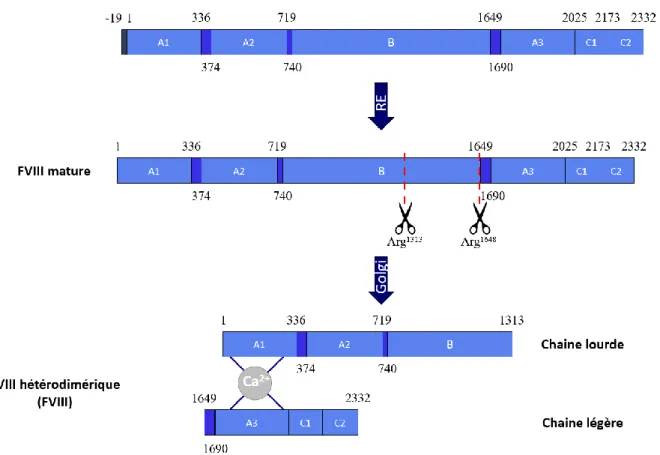
Synthèse du FVIII
La séquence signal permet l’adressage à la lumière du réticulum endoplasmique où elle est clivée, donnant la protéine mature de 2332 aa. C’est dans le RE que le FVIII subit des N-glycosylations et interagit avec la calréticuline et la calnexine36. Une large proportion
(80%) interagit également avec la protéine BiP via le domaine A1, qui le retient dans le RE pour dégradation37,38. Le reste passe dans l’appareil de Golgi de manière
ATP-dépendante où la maturation du FVIII se poursuit. Dans le Golgi, les N-glycosylations se complexifient, des O-glycosylations et des sulfatations apparaissent. C’est également le lieu des premières protéolyses menant au FVIII fonctionnel.
En effet, le FVIII est clivé par une (des) protéase(s) intracellulaire(s) au niveau de l’arginine 1613 (Arg1313) et de l’arginine 1648 (Arg1648). Si la protéase clivant en position
1648 a été décrite comme étant la furine39,40, son identité reste malgré tout
controversée41. Quoi qu’il en soit, ce clivage en fin de domaine B aboutit à la séparation de
la chaine lourde (A1-a1-A2-a2-B) d’une part et de la chaine légère (a3-A3-C1-C2) d’autre part. Ces 2 chaines sont reliées de manière non covalente par les domaines A1 et A3, par l’intermédiaire d’ions métalliques divalents, notamment le calcium, le cuivre ou le manganèse42,43. Cependant, il semble que les ions cuivre présents dans molécule de FVIII
dans l’association de ses deux chaines44. C’est sous cette forme hétérodimérique inactive
qu’est sécrété le FVIII et qu’il circule dans le plasma à environ 0,1 µg/mL.
Figure 2. Clivage intracellulaire du FVIII. Dans le réticulum endoplasmique, la séquence signal est clivée et le polypeptide de 2332 aa est clivé au niveau des Arg1313 et Arg1648 dans l’appareil de Golgi. C’est également dans l’appareil de Golgi que le FVIII subit des modifications post-traductionnelles, comme des O-glycosylations, des N-glycosylations et des sulfatations de tyrosines spécifiques. Le FVIII est alors sous forme hétérodimérique, avec une chaine lourde (domaines A1-a1-A2-a2-B) et une chaine légère (domaines a3-A3-C1-C2). Les deux chaines sont associées de manière non covalente par un ion divalent entre les domaines A1 et A3. Il est sécrété sous cette forme dans le sang et interagit immédiatement avec le vWF.
Demi-vie du FVIII
Immédiatement après sa sécrétion dans la circulation, le FVIII se fixe à sa protéine chaperonne, le vWF. Il a en effet été déterminé que 50% du FVIII est complexé au vWF en seulement 2 secondes45. De plus, il se fixe environ 50 molécules de vWF par molécule de
FVIII et cette interaction est caractérisée par une forte affinité (entre 0.1 et 0.9 nM), mais également une dissociation très rapide (6,89.10-3 s-1)46. Cette interaction se fait via les
deux extrémités de la chaine légère, à la position Tyr1680 préalablement sulfatée dans le
Willebrand est de protéger le FVIII d’une inactivation prématurée du FVIII par les protéases et permet qu’il ne s’active qu’à proximité d’une lésion vasculaire jusqu’où il l’escorte.
Par ailleurs, le vWF évite également une reconnaissance par les récepteurs impliqués dans le catabolisme du FVIII et augmente ainsi considérablement sa demi-vie. En effet, chez les individus ayant des taux normaux de vWF, la demi-vie du FVIII est d’environ 12 heures51. En revanche, chez les patients atteints de déficiences en vWF (maladie de
Willebrand de type 3), elle est de 2,8 heures en moyenne52. Il a d’ailleurs été montré que
chez les patients hémophiles A, 0,1 unité/mL de vWF en plus augmente de 17 minutes la demi-vie du FVIII53. Cependant, le vWF circule en excès par rapport au FVIII, ce qui signifie
que cet effet est vrai à faible dose de vWF54. En revanche, la pertinence de cette
observation chez les patients avec des taux normaux de vWF est limitée, comme en témoigne la moindre augmentation dans une cohorte de plus de 11 000 individus55.
Enfin, nous verrons dans les chapitres suivants que le vWF joue également un rôle prépondérant quant à l’immunogénicité du FVIII.
Rôle du FVIII
Lorsqu’une brèche vasculaire apparait, elle expose le sous-endothélium et sa matrice de collagène à la circulation. Grâce à une vasoconstriction concomitante, les plaquettes sont ralenties et adhèrent à la matrice, s’agrègent et s’activent pour recruter encore plus de plaquettes et ainsi aboutir à la formation d’un clou plaquettaire. L’ensemble de ce processus correspond à l’hémostase primaire et donne lieu à la formation d’un clou hémostatique. Chez les patients hémophiles A, l’hémostase primaire est normale. Cependant, la présence de ce clou ne suffit pas à collapser la brèche pour une bonne coagulation. Il doit être solidifié pour être stabilisé et c’est à cette fin qu’est activée la cascade de la coagulation.
La cascade de la coagulation consiste en l’activation séquentielle de zymogènes (précurseurs enzymatiques inactifs) par l’enzyme présente en amont dans la cascade. Ces enzymes, appelées « facteurs » sont des sérines protéases qui doivent être clivées pour devenir actives. Une fois actives, elles clivent à leur tour le facteur suivant grâce et leur
cofacteur glycoprotéique qui catalyse la réaction. La cascade de la coagulation débute de deux façons distinctes : par la voie extrinsèque et par la voie intrinsèque.
La voie extrinsèque se déclenche via le facteur tissulaire (FT) exposé, en plus du collagène, lors de la lésion vasculaire. Ce FT active le Facteur VII présent dans le plasma en facteur VII activé (FVII → FVIIa). Le complexe FT-FVIIa active alors le Facteur X (FX → FXa). Le FXa génère un peu de FVa56 auquel il se lie pour former le complexe prothrombinase, clive
la prothrombine et permet la génération des premières molécules de thrombine. Cette thrombine clive le FVIII au niveau de l’Arg372, l’Arg740 et l’Arg1689 avec pour conséquence
son activation (FVIII → FVIIIa)57.
Figure 3. Activation du FVIII. Lors de la cascade de la coagulation, la thrombine active le FVIII en le clivant au niveau de la chaine lourde (Arg372 et Arg740) et de la chaine légère (Arg1689) pour générer FVIII actif (FVIIIa). Les domaines a1 et A2 interagissent alors par des interactions faibles (││││││││││││││││), le FVIII est alors amputé du domaine B et libéré du vWF. Il peut se lier aux
phospholipides via son domaine C2 et interagit avec le FIXa via ses domaines A2 et A3. En bas à droite : Structure cristallographique du FVIII (D’après van den Biggelaar et al., J. Biol. Chem., 2015).
En parallèle, la voie intrinsèque s’initie par la fixation du Facteur XII sur des surfaces chargées négativement du sous-endothélium. Ce FXII s’auto-active (FXII → FXIIa) et active
le Facteur XI (FXI → FXIa). Ce dernier active à son tour le Facteur IX (FIX → FIXa). Le FIXa s’associe alors à son cofacteur, le FVIIIa en présence de calcium et de phosphatidylsérine (PS) des plaquettes activées pour former le complexe ténase. Ce dernier se lie alors à son substrat, le Facteur X pour l’activer (FX → FXa). Le FXa se trouve alors à la jonction des 2 voies d’activation et son importante génération entraine la génération de thrombine. La thrombine transforme le fibrinogène en fibrine d’une part et active le Facteur XIII (FXIII → FXIIIa), aboutissant à la stabilisation du caillot de fibrine par le FXIIIa58. Par ailleurs, la
génération de thrombine sert également d’activation de voies d’amplification puisque la thrombine active le FVIII, nous l’avons vu, mais également le FV ; tous deux nécessaires à sa propre production.
Figure 4. Cascade de la coagulation. L’exposition du facteur tissulaire (FT) et du collagène déclenche simultanément la voie extrinsèque et la voie intrinsèque de la cascade de la coagulation, respectivement. Le FT active le facteur VII (FVII FVIIa) et se lie au FVIIa pour activer le facteur
X (FX FXa). Le FXa génère de petites quantités de facteur V activé (FVa), permettant la génération précoce de thrombine en clivant la prothrombine. Cette thrombine clive le facteur VIII et l’active (FVIII FVIIIa). En parallèle, le facteur XII s’auto-active au contact du sous-endothélium exposé (FXII FXIIa) et active le facteur XI (FXI FXIa). Ce dernier active le facteur IX (FIX FIXa) qui s’associe au FVIIIa pour activer son substrat, le FX et générer de grandes quantités de thrombine. La thrombine transforme alors le fibrinogène en fibrine et active le facteur XIII (FXIII FXIIIa) qui stabilise le caillot de fibrine.
Catabolisme du FVIII
Le récepteur LRP1 (Lipoprotein receptor-related protein-1) ou CD91 est un récepteur d’endocytose connu pour son rôle de maintien de l’homéostasie, notamment du cholestérol59. Or, il s’avère que LRP1 reconnait également le FVIII in vitro et permet sa
dégradation intracellulaire60. Ainsi, in vivo une déficience en LRP1 se traduit par un temps
de présence du FVIII dans le plasma 1,5 fois plus importante61. Selon le modèle proposé
par Sarafanov et Saenko, le catabolisme du FVIII via LRP1 se ferait en deux étapes62,63. La
première consiste en la fixation et l’accumulation de FVIII à la surface de la cellule après liaison entre son domaine A2 et des protéoglycanes héparanes sulfates (heparan sulfate proteoglycan - HSPG). A ce stade, le FVIII est complexé au vWF, qui se détache, révélant un site de liaison du LRP1 sur le domaine C2 du FVIII. C’est suite à cette reconnaissance du domaine C2 que le FVIII est endocyté et dégradé. Cette hypothèse est étayée in vivo par le fait que le blocage simultané des HSPG et de LRP1 aboutit à une augmentation de la demi-vie du FVIII de 5,5 fois.
Les études précédemment citées ont utilisé la molécule RAP pour inhiber LRP1 et démontrent une diminution plus importante du catabolisme que les modèles déficients en LRP1. Or, RAP n’est pas un antagoniste exclusif du LRP1, mais il inhibe l’ensemble des récepteurs aux lipoprotéines de faible densité (low density lipoprotein – LDL). On peut donc supposer que cette différence est due à l’intervention d’autres récepteurs aux LDL dans le catabolisme du FVIII. C’est ce qu’ont confirmé les travaux d’Ananyeva et al. in
vitro64 et ceux de Bovenschen et al. in vivo65.
Il semble par ailleurs que le FVIII soit également reconnu par ses motifs post-traductionnels. Ainsi, le domaine B du FVIII, très fortement N-glycosylé, est-il reconnu par l’Asialoglycoprotein Receptor (ASGPR), une lectine de type C66.
II. Traitements de l’Hémophilie A et options thérapeutiques
1. Traitements actuelsFVIII thérapeutiques
A l’heure actuelle, le traitement des patients atteints d’hémophilie A consiste en l’injection intraveineuse de FVIII exogène afin de restaurer des taux suffisants de FVIII en circulation. L’administration de ce FVIII dit « thérapeutique » permet de prévenir et/ou de traiter les hémorragies, puisqu’elle se fait à la demande (à la suite d’un épisode hémorragique) mais surtout en prophylaxie (en prévention de l’apparition d’hémorragie).
Lorsqu’il est utilisé en prophylaxie, pour prévenir l’apparition de symptômes tels que l’arthropathie, le FVIII thérapeutique est injecté 2 à 3 fois par semaine à faible dose, soit 25 à 40 UI/kg67,68. En effet, l’objectif ici est de modifier le phénotype hémorragique de
sévère à modéré, puisqu’il a été montré une protection contre les symptômes les plus lourds et donc une meilleure qualité de vie chez les patients hémophiles A modérés69,70.
Cet effet protecteur contre les hémarthroses et autres saignements à 25 UI/kg a été confirmé chez des patients71. En revanche, l’adhésion du patient est critique pour obtenir
une bonne observance du traitement, surtout chez les jeunes enfants, ce qui constitue une limite importante à cette stratégie et pose la question de l’âge de début de la prophylaxie72. En ce qui concerne la prise en charge à la demande, les protocoles sont plus
variables selon l’intensité du saignement mais les taux de FVIII à atteindre sont beaucoup plus importants (40-100% d’activité normale du FVIII)67.
Dans les pays dits « développés », le FVIII thérapeutique provient de deux sources : dérivé de plasma (dérivés plasmatiques ou « plasma derived FVIII » - pdFVIII) ou issu du génie génétique (recombinant - rFVIII). Le pdFVIII est un FVIII précipité puis purifié par chromatographie à partir d’un mélange de plasma de donneurs sains73. Depuis les années
1980 et le scandale du sang contaminé, au cours duquel 60 à 70% des patients hémophiles ont été contaminés par le virus du SIDA (VIH) et celui de l’hépatite C (VHC), les pdFVIII subissent également des étapes d’inactivations virales et le risque infectieux à quasiment disparu74. Une attention particulière doit cependant être portée sur la transmission de
protéines présentes dans les préparations de pdFVIII correspondent à d’autres protéines purifiées avec le FVIII telles que le vWF, l’albumine ou la fibronectine76. Cela pose
problème quant à l’immunogénicité de ces préparations77. L’ensemble de ces limitations
a favorisé l’essor de la production de FVIII recombinants. Ces FVIII sont synthétisés à partir de la séquence ADN de deux donneurs78 et produits dans des lignées cellulaires de
hamster. Baxter/Shire utilise des cellules CHO (Chinese Hamster Ovary) pour produire l’Advate®, tandis que Bayer et CSL-Behring utilisent des cellules BHK (Baby Hamster
Kidney) pour le Kogenate® et l’Helixate®, respectivement. Plus récemment, la production
de ce rFVIII a été optimisée par Wyeth/Pfizer en synthétisant un FVIII directement sans domaine B (B Domain Deleted FVIII - BDD FVIII), le Refacto® et Octapharma a produit son
FVIII BDD, le Nuwiq® dans des cellules humaines, les HEK293 (Human Embryonic
Kidney)79.
Thérapies sans FVIII
Cependant, le FVIII, de par sa taille, reste difficile et donc couteux à produire. C’est une des raisons pour laquelle d’autres axes de recherche alternatifs à l’utilisation de FVIII ont été développés.
Le premier d’entre eux vise à court-circuiter le FVIII dans la cascade de la coagulation (« by-passing agents »). On trouve ainsi l’utilisation de FVIIa, qui permet la génération de thrombine par la voie extrinsèque80, ou encore de concentrés de protéines du complexe
prothrombinique activé (activated Prothrombin Complex Concentrates – aPCC)81. Les
aPCC, dont le FEIBA® de Baxter est le seul commercialisé, comprennent toutes les
enzymes et cofacteurs nécessaires à la génération de thrombine (prothrombine, FVIIa, le FIXa, le FXa…).
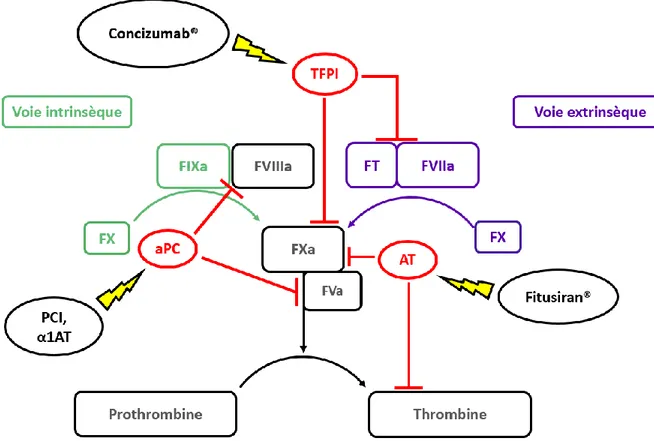
La deuxième stratégie se propose de rétablir la coagulation en inhibant les régulateurs naturels de celle-ci. Ainsi, la protéine C activée (activated protein C - aPC) inactive le FV et le FVIII57,82. C’est pourquoi plusieurs molécules comme l’inhibiteur de la protéine C
(protein C inhibitor -PCI) et l’α1-antitrypsin (α1AT) sont utilisées afin de favoriser la coagulation83,84. De façon similaire, la kinase Tissue Factor Pathway Inhibitor (TFPI)
régule la coagulation en inhibant non seulement le FXa, mais également le complexe TF-FVIIa85. C’est pourquoi cette molécule a été choisie comme cible pour être inhibée par un
anticorps monoclonal, le Concizumab®86. Cet anticorps inhibe la liaison du TFPI au FXa et
empêche également l’inhibition du complexe FT-FVIIa, permettant ainsi le rétablissement de la coagulation87,88. L’antithrombine (AT) elle, exerce son effet anticoagulant en
inhibant la thrombine et dans une moindre mesure le FIXa89. Ainsi, le Fitusiran®, un petit
ARN interférant (small interfering RNA – siRNA) développé par Alnylam, inhibe la traduction de l’ARNm codant l’AT et favorise donc la coagulation90.
Figure 5. Régulateurs naturels de la cascade de la coagulation et stratégies pour les inhiber. La kinase Tissue Factor Pathway Inhibitor (TFPI) inhibe le FXa et le complexe FT-FVIIa. Le Concizumab® est un anticorps anti-TFPI monoclonal thérapeutique qui inhibe la liaison du TFPI à ces cibles. De même, l’antithrombine (AT) inhibe le complexe FT-FVIIa et dans une moindre mesure le FXa. Le Fitusiran® est un petit ARN interférant qui inhibe la synthèse de l’AT. Enfin, la protéine C activée (aPC) inactive le FVa et le FVIIIa. L’inhibiteur de la protéine C (PCI) et l’α1-antitrypsin (α1AT) inhibent l’action de l’aPC.
2. Nouvelles stratégies
Si les patients hémophiles sont à 87% « satisfaits / plutôt satisfaits » de leur traitement actuel, 55% d’entre eux espèrent que les nouveaux traitements leur permettront de diminuer la fréquence des injections91. Cette étude, menée sur plus de 1 000 patients
hémophiles (dont 85 % atteints d’hémophilie A) entre janvier 2015 et août 2016 est très intéressante car elle compile les attentes des patients quant aux futurs traitements. Il en ressort que ces attentes se regroupent en deux catégories : tout d’abord la qualité de vie (fréquence d’injection, problème d’accès aux veines, stockage, flexibilité de gestion du traitement…) et ensuite la sécurité du traitement (absence d’effets secondaires, peur de l’apparition d’une réponse immunitaire…). A noter que cette étude a été faite en Allemagne, Suisse et Autriche, pays où l’accès aux centres de soins et aux traitements n’est pas représentatif de la situation mondiale. Cela explique probablement que la baisse du prix du traitement, bien qu’estimé à plus de 50 000€/an/patient92,93, ne constitue une
attente que pour 5% des patients interrogés.
FVIII à longue durée de vie
En réponse à ces attentes et ainsi augmenter l’adhésion aux traitements, de nouveaux traitements ont été développés. La première évolution a été la mise sur le marché de FVIII à longue durée de vie afin de diminuer la fréquence des injections et de protéger les articulations des patients en favorisant une présence constante de FVIII dans le sang. Pour augmenter la demi-vie du FVIII, deux stratégies sont actuellement utilisées. La première d’entre elles consiste à fusionner la région constante (partie Fc) de l’IgG humaine à l’extrémité C-terminale du FVIII. Les IgG circulent en effet pendant environ 3 semaines dans le corps, notamment grâce à leur liaison au récepteur Fc néonatal (FcRn) qui les protège de la dégradation lysosomale94. La fusion de la partie Fc vise donc à utiliser ce
processus biologique afin de préserver le FVIII de la dégradation et ainsi augmenter sa demi-vie. Ainsi, l’Eloctate® développé par Bioverativ montre-t-il une demi-vie 1,5 fois plus
longue qu’un BDD normal95. Ce produit a d’ailleurs fait l’objet d’un essai clinique de phase
III (Numéro ClinicalTrials.gov : NCT01454739) qui semble valider son effet et son innocuité à long terme96.
La seconde stratégie pour allonger la demi-vie du FVIII thérapeutique repose sur sa PEGylation, qui consiste en la liaison au FVIII de polymères de polyéthylène glycol (PEG). Cette PEGylation permet d’une part d’augmenter la taille des molécules thérapeutiques et ainsi diminuer la clairance rénale, et d'autre part de les protéger des protéases et de l’endocytose par les cellules « scavenger » comme les macrophages ou les cellules
endothéliales97. Cela résulte donc en une augmentation de la demi-vie, de 1,6 fois dans le
cas du FVIII98. A nouveau, des essais cliniques de l’Adinovate® de Shire (Numéro
ClinicalTrials.gov : NCT01736475) ou du N8-GP de NovoNordisk ont confirmé l’allongement de la demi-vie et la sureté des produits99.
Par ailleurs, d’autres stratégies sont actuellement en développement pour augmenter la demi-vie du FVIII, comme par exemple la synthèse d’un FVIII monocaténaire (single-chain FVIII – scFVIII) avec une affinité environ deux fois supérieure pour le vWF que le FVIII, in
vivo100. Cependant, l’amélioration de demi-vie obtenue est pour l’instant plus faible
qu’avec les stratégies précédemment citées, puisqu’elle est de l’ordre de 1,2 fois101.
Certaines de ces stratégies ont également été utilisées pour prolonger la demi-vie du FIX et permettent une augmentation de 4 à 6 fois de sa demi-vie. L’écart observé entre le gain pour le FVIII et le FIX s’explique par le fait que le catabolisme du FVIII est grandement régulé par son interaction avec sa protéine chaperonne. Nous l’avons vu, en l’absence de vWF, la demi-vie du FVIII est presque 6 fois plus courte. Ainsi, le facteur limitant l’augmentation de la demi-vie du FVIII est la demi-vie du vWF (approximativement 15h). Le FVIII, qu’il ait une demi-vie prolongée ou pas, est dégradé en étant complexé avec le vWF. A l’inverse, le FIX n’a pas cette limitation et lors d’un essai clinique en 2014, il a par exemple été calculé pour le FIX-Fc, une demi-vie de 82h, soit 5 fois supérieure à celle du FIX102–104.
Figure 6. FVIII à longue durée de vie et essais cliniques correspondants. D’après Mancuso et Santagostino, J. Clin. Med., 2017
Emicizumab
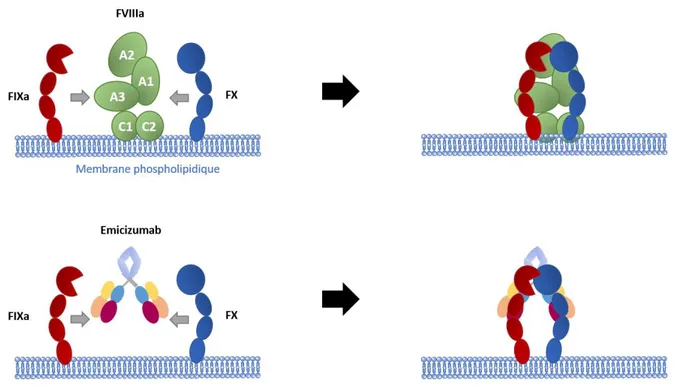
Une nouvelle molécule est, elle, déjà approuvée par l’agence américaine en charge des médicaments (Food and Drugs Administration – FDA) sous le nom d’Hemlibra®
(Roche)105. Cette molécule, l’Emicizumab, remplace le FVIII sans pour autant être du FVIII.
Il s’agit en effet d’un anticorps bispécifique, qui reconnait avec l’un de ses fragments de liaison à l’antigène (antigen-binding fragment – fab) le FIXa et avec le second fab, le FX106.
Cette double reconnaissance permet le rapprochement spatial des deux molécules, permettant ainsi à l’enzyme (le FIXa) d’activer son substrat (le FX) sans l’intervention de FVIII107. Le premier essai clinique mené chez l’homme a montré une demi-vie de 4 à 5
semaines après une injection sous-cutanée d’Emicizumab108. Les essais cliniques suivants
ont par la suite démontré une forte diminution du nombre de saignements annuels (Annual Bleeding Rate – ABR), voire une disparition totale des saignements chez plus de 70% des patients109. Un programme d’essais cliniques nommé HAVEN a depuis vu le jour
et les 4 essais cliniques qu’il comprend confirment une correction du phénotype hémorragique, particulièrement lorsqu’il est donné en prophylaxie110. Cependant, des
micro-angiopathies thrombotiques ont été décrites chez certains patients traités en parallèle avec des aPCC. Cela est probablement dû au fait que les aPCC contiennent du
FIXa et du FX, comme décrit précédemment (paragraphe « Thérapies sans FVIII »), élément essentiel de l’action de l’Emicizumab et entrainant donc des symptômes thrombotiques111. Ainsi, il est recommandé d’éviter de traiter les patients avec des aPCC
lorsqu’ils utilisent l’Emicizumab en prophylaxie. Il est plutôt conseillé de privilégier du FVIIa pour lequel aucune thrombose n’a été décrite112.
Par ailleurs, la particularité de cette très élégante stratégie requiert une attention particulière quant à son monitorage. En effet, l’Emicizumab est très différent d’un point de vue moléculaire du FVIII qu’il remplace et cela entraine un grand nombre de conséquences. En plus des sites de fixation et de la spécificité qui sont différents entre les deux molécules, la pharmacocinétique varie également beaucoup. Ainsi, si les niveaux d’Emicizumab dans le plasma sont estimés comme équivalents à 10 à 15% de l’activité hémostatique normale du FVIII113, la plus faible affinité de l’anticorps bispécifique pour
ses ligands entraine la formation de moins de complexe ténase que pour une concentration en FVIII équivalente114. Enfin, le FVIII nécessite une activation préalable
pour jouer son rôle de cofacteur et peut être inactivé (par l’aPC notamment) alors que l’Emicizumab n’est sensible à aucun de ces mécanismes de régulation.
Figure 7. Schéma du mode d’action du FVIIIa ou de l’Emicizumab. La double reconnaissance du FIXa et du FX permet le rapprochement physique de l’enzyme et du substrat, permettant une activation du FX en FXa. D’après Kitazawa, Nat. Med., 2012.
Thérapie génique
Enfin, la troisième stratégie a pour objectif de guérir l’hémophilie A. Il s’agit de la thérapie génique. Cette stratégie vise à incorporer dans les cellules du patient une version saine du gène provoquant la maladie, permettant théoriquement une cure du patient. Le caractère monogénique de l’hémophilie (causée par un seul gène) et le fait qu’une faible augmentation de l’activité pro-coagulante permette une nette amélioration du phénotype hémorragique font de l’hémophilie un candidat idéal à la thérapie génique, comme en témoignent les avancées majeures obtenues pour l’hémophilie B115,116. En ce qui concerne
l’hémophilie A, les contraintes sont plus importantes puisqu’il se rajoute, aux difficultés intrinsèques à la thérapie génique (expression du transgène, immunogénicité…), la très grande taille du gène F8 et de son transcrit. Les éléments essentiels à prendre en compte sont donc : le vecteur utilisé, le tissu cible et le gène du FVIII.
Ainsi la recherche sur les vecteurs viraux nécessaires au transport du gène et à son intégration, a-t-elle été fondamentale au développement de la thérapie génique pour l’hémophilie A. Les vecteurs sont des acides nucléiques viraux (ADN ou ARN) qui sont utilisés pour tirer avantage de la capacité naturelle des virus à infecter une cellule cible et à utiliser la machinerie cellulaire pour leur propre réplication. Ainsi, les vecteurs les plus couramment utilisés sont les lentivirus (génome à ARN) et les AAV (Adeno-Associated Virus, génome à ADN). Si les lentivirus semblent plus adaptés pour l’hémophilie A car de plus grands gènes peuvent y être insérés117, les AAV ont l’avantage d’être moins
immunogènes et non intégratifs, limitant ainsi le risque de mutation intégrationnelle118.
En effet, l’immunogénicité et la toxicité (des vecteurs, de la protéine synthétisée) représentent de grandes contraintes en thérapie génique. C’est pourquoi des AAV recombinants ont été développés à partir des nombreux sérotypes existants naturellement, offrant ainsi aux chercheurs une boite à outils d’AAV très diverse119. Par
ailleurs, les différents AAV présentent des tropismes pour certains organes et vont donc permettre une expression préférentiellement dans ceux-ci120.
C’est également par le choix du promoteur du gène d’intérêt que l’on peut cibler un organe. En effet, le choix du promoteur et de ses caractéristiques est une étape essentielle du développement d’une thérapie génique. Un promoteur peut être choisi pour sa
spécificité d’expression (exprimé dans un certain type cellulaire ou tissu) ou pour sa « force » (sa capacité à promouvoir l’expression du gène qui le suit).
En parallèle, le gène du FVIII a lui aussi subi un grand nombre d’améliorations. Non seulement pour en diminuer la taille (utilisation d’un gène codant un FVIII BDD), mais également pour augmenter son taux de synthèse par exemple en faisant de la codon-optimisation. La codon-optimisation consiste en effet à choisir les codons pour lesquels les ARN de transfert (ARNt) sont les plus fréquents chez l’hôte et ainsi permettre une augmentation du taux de traduction121.
L’ensemble de ces développements a permis le lancement de plusieurs essais cliniques qui n’ont pas permis d’aboutir à une correction phénotypique à long terme121. Cependant,
un nouvel essai clinique de thérapie génique a été lancé en aout 2015 (Numéro ClinicalTrials.gov : NCT02576795). Cet essai de phases I/II vise à évaluer l’efficacité du Valoctocogene Roxaparvovec® (BMN 270 - Biomarin Pharmaceutical Inc.), une thérapie
AAV5 codant pour un FVIII BDD codon-optimisé, sous le contrôle d’un promoteur spécifique du foie122. Si les résultats finals sont attendus pour 2022, des résultats
préliminaires sont déjà parus. Parmi les 7 patients traités avec la plus forte dose (6.1013
vg/kg), 5 ont des taux de FVIII entre 8 et 60% de l’activité normale, entre 5 et 16 semaines post-injection123. Après 20 semaines, les taux de FVIII sont supérieurs à 50% pour tous
les patients sauf un (entre 12 et 32%). Par ailleurs, il a été constaté une augmentation de la cytotoxicité hépatique (via les taux d’Aspartate AminoTransférase -ASAT- et d’Alanine AminoTransférase -ALAT-) chez 8 patients sur les 9 compris dans l’essai clinique. Cependant, chez un patient seulement, l’augmentation d’ALAT s’est accompagnée d’une diminution de l’activité du FVIII, ce qui a été résolu par l’utilisation de glucocorticoïdes124.
Depuis, d’autres essais cliniques ont été lancés et on en dénombre aujourd’hui sept autres en phase de recrutement (www.clinicaltrials.gov, recherche « hemophilia A » + « gene therapy »).
Cependant, ces essais cliniques risquent de se confronter à la préexistence, chez 15 à 20% de la population, d’anticorps anti-AAV125 à des doses qui, bien que faibles, bloquent
complètement la transduction des AAV126. Chez les patients hémophiles, ces titres
d’anticorps sont encore plus élevés et atteignent 43,5% des patients pour certains sérotypes127. En effet, il a été montré que des cellules de l’immunité innée reconnaissent
donné la forte homologie entre les différents sérotypes, tous sont reconnus, inégalement, par ces anticorps. Dans l’essai clinique cité précédemment, des anticorps anti-AAV ont d’ailleurs été détectés chez tous les participants dès la huitième semaine post-injection124.
De nombreuses stratégies pour surmonter cette difficulté sont imaginées, notamment l’utilisation d’immunosuppresseurs, mais toutes présentent des inconvénients126. La
thérapie génique est donc, dans le cadre de l’hémophilie A, très prometteuse, sans pour autant pouvoir proposer une solution à court terme pour l’ensemble des patients.
Le premier projet de cette thèse vise à tester une nouvelle stratégie thérapeutique intermédiaire entre la thérapie substitutive et la thérapie génique. Cette stratégie repose sur l’injection d’ARNm codant le FVIII.
III. Complication du traitement
1. Immunogénicité du FVIII thérapeutique
Lors d’un traitement de substitution avec du FVIII, la complication la plus redoutée à l’heure actuelle est l’apparition d’une réponse immunitaire anti-FVIII. En effet, chez 25 à 30% des patients hémophiles A sévères traités avec du FVIII thérapeutique, on constate l’apparition d’anticorps anti-FVIII qui inhibent son activité pro-coagulante et sont donc appelés « inhibiteurs »128–130. Le traitement perd alors son efficacité et les patients font
face à une impasse thérapeutique. Il est intéressant de noter que l’on constate également le développement d’inhibiteurs chez 7% des patients atteints d’une hémophilie A non sévère après 50 jours d’exposition131.
L’apparition d’inhibiteurs est donc redoutée par les cliniciens car elle représente, nous l’avons vu, une impasse thérapeutique, mais elle constitue aussi la 3e préoccupation
majeure des patients91. En effet, en plus de voir leur qualité de vie se dégrader, leur taux
de mortalité a longtemps augmenté, allant jusqu’à doubler avant 1992132. Aujourd’hui, si
le taux de morbidité est toujours augmenté, l’apparition d’inhibiteurs ne semble plus significativement augmenter la mortalité. Cela se fait cependant au prix d’un triplement du coût de prise en charge, avec un coût en 2003 dépassant les 200 000 euros par an et
L’identification des facteurs et la compréhension des mécanismes menant à l’apparition d’inhibiteurs est indispensable pour, à terme, améliorer la qualité de vie des patients hémophiles. Ainsi, de nombreuses études ont été menées pour identifier de potentiels facteurs prédictifs et tenter de prévenir l’apparition d’une réponse immunitaire anti-FVIII. Ces facteurs ont été classés en deux catégories : les facteurs liés aux patients (intrinsèques) et ceux indépendants des patients (environnementaux).
2. Facteurs de risques intrinsèques aux patients
Le fait que de si nombreuses mutations du gène codant le FVIII aient été décrites peut s’expliquer par la localisation du gène F8. Celui-ci se situe dans une région constituée de nombreuses séquences répétées, ce qui se traduit par un important taux de mutations (3,4.10-5 mutations/division cellulaire pour F8 vs 1.10-6
mutations/gène/division cellulaire en moyenne)133. Ainsi, le facteur génétique apparait
comme évident, d’autant plus si l’on considère que les patients avec un antécédent familial d’inhibiteurs ont trois fois plus de risque d’en développer également134.
Anomalies du gène codant le FVIII
Le type de mutation responsable de l’hémophilie A a été identifié comme facteur de risque. Les patients ayant une large délétion du gène F8 ou une mutation non-sens ont plus de risque de développer des inhibiteurs que ceux porteurs d’une inversion de l’intron 2222. De même, les patients hémophiles A sévères sont 6 fois plus nombreux (30% vs 5%)
que les hémophiles A modérés à développer des inhibiteurs135. Cependant, il existe des
patients dits « CRM négatifs » (Cross-Reacting Material) qui n’ont pas d’antigène FVIII détectable dans le plasma. On pourrait alors s’attendre à ce que la tolérance centrale au FVIII soit impossible et que pour le système immunitaire de ces patients le FVIII thérapeutique soit considéré comme du non-Soi et qu’ils déclenchent donc systématiquement une réponse immunitaire. Or, tous ne le font pas avec, par exemple, seulement environ 20% de patients avec une inversion de l’intron 22 qui développent des inhibiteurs, malgré leur phénotype CRM-négatif136. Ce genre d’observation amène à
penser que si la mutation du gène F8 est très importante, voire prépondérante dans le développement d’inhibiteurs, elle est néanmoins insuffisante à l’expliquer137.
Autres gènes
Des études chez le patient ont permis d’identifier certains allèles du complexe majeur d'histocompatibilité de classe II (CMH II) nommés HLA (human leukocyte antigen) comme facteur de risque138,139. Ces observations ont par la suite été confirmées in
silico140,141 et pourraient signifier que certains patients CRM négatifs synthétisent des
parties du FVIII qui sont capables d’engendrer un complexe CMH-peptide suffisamment stable pour induire une tolérance centrale.
Parallèlement, de nombreux autres polymorphismes ont été décrits comme ayant un rôle dans le développement ou la protection contre les inhibiteurs. Il s’agit de polymorphismes dans des gènes codant pour des protéines de l’immunité. On trouve notamment des gènes codant des interleukines (IL), telles que l’IL-1α, l’IL-1β, l’IL-2 ou l’IL-12142. Cependant,
l’identification de facteurs de risque/de protection se heurte aux faibles effectifs des cohortes de patients hémophiles A et d’autres études nuancent ces résultats134,143. Mais le
polymorphisme le plus couramment décrit est celui dans le promoteur du gène codant l’IL-10137,144. Cela peut s’expliquer par le fait que l’IL-10 est un médiateur immunitaire
crucial, notamment pour la prolifération des lymphocytes T (LT) régulateurs (Treg), cellules essentielles à la régulation de la réponse immunitaire145. Des polymorphismes
dans les promoteurs des gènes codant pour le TNFα (Tumor Necrosis Factor α), CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4) et l’hème oxygénase (HOX-1) ont également été identifiés146–148. Enfin, il a été récemment montré que le groupe sanguin O est associé à
une diminution du risque d’approximativement deux fois, bien que les raisons de cette protection restent peu claires149.
Ethnie
D’autres facteurs liés au patient ont également été décrits. Parmi eux, l’ethnie d’origine joue un rôle dans la probabilité de développer des inhibiteurs. En effet, il semble que les patients non caucasiens (hispaniques, africains, asiatiques, indiens) aient jusqu’à 5 fois plus de risque de développer des inhibiteurs150–154. Cela peut être relié aux séquences
ADN (de deux donneurs caucasiens) à partir desquelles les FVIII recombinants ont été clonés155, ou à la différente répartition des allèles HLA selon les ethnies. Cependant, cette
3. Autres facteurs de risques
Si l’étude de cohortes et de familles avec un historique d’inhibiteurs a permis de confirmer la forte implication de l’aspect génétique, seule la mutation de F8 ou les antécédents semblent systématiquement déterminants quant à l’apparition d’inhibiteurs. Ainsi, il semble clair désormais que l’apparition de ces anticorps est liée à une interaction complexe entre des facteurs intrinsèques au patient et à des facteurs qui lui sont externes. Ces facteurs regroupent l’ensemble des variables reliées au traitement du patient. C’est pourquoi ils sont d’autant plus importants et intéressants, puisqu’ils représentent le levier permettant aux cliniciens d’agir afin de diminuer la probabilité d’apparition des inhibiteurs.
Antécédent de traitement
La première chose à prendre en compte pour monter des études sur les facteurs de risque indépendants du patient est de distinguer ceux ayant déjà reçu du FVIII (previously treated patients – PTPs) ou pas (previously untreated patients – PUPs). En effet, il a été montré que les PUPs ont une incidence d’inhibiteurs 30 fois plus importante que les PTPs156. De plus, les études menées sur les PUPs permettent d’éviter les biais liés aux
traitements précédents. C’est pourquoi ce groupe de patients est le plus utilisé pour ces analyses157.
Le FVIII lui-même
La molécule de FVIII elle-même est une source d’immunogénicité. En effet, les mécanismes d’endocytose seront détaillés plus loin, mais la reconnaissance du FVIII par les cellules immunitaires est un élément primordial dans le déclenchement de la réponse immunitaire. Des motifs comme les glycanes se terminant par des mannoses, ont été identifiés comme essentiels à la reconnaissance des pathogènes, notamment microbiens. Dans le cadre du FVIII, la présence d’un glycan se terminant par du mannose en position 2118 a été montrée comme favorisant l’endocytose du FVIII par des cellules dendritiques dérivées de monocytes humains (MO-DC) via le récepteur CD206 in vitro158,159. De même,
ces anticorps, que certains domaines du FVIII favorisent sa reconnaissance. En effet, in
vitro certains résidus hydrophobes du domaine C2 favorisent son endocytose et la
présentation aux LT160. In vivo, le domaine C1 joue également un rôle dans
l’immunogénicité du FVIII puisque la triple mutation R2090A/K2092A/F2093A réduit l’activation des LT spécifiques du FVIII161,162.
Plusieurs études ont montré que la structure du FVIII ne permettait pas une maturation des cellules dendritiques, impliquées dans le déclenchement des réponses immunitaires163,164. Dès lors, outre la structure du FVIII, sa fonction a également été
évoquée comme facteur de risque. Il a ainsi été proposé que la génération de thrombine induite après injection de FVIII thérapeutique puisse constituer un signal de co-stimulation qui, ajouté à une reconnaissance du FVIII par les cellules immunitaires, engendre le développement de la réponse immunitaire165. Cependant, une autre étude
visant à étudier l’influence de l’explosion de génération de thrombine sur le développement d’inhibiteurs a remis en cause ces résultats. En effet, dans cette étude, ni l’injection d’un FVIII sans activité, ni le blocage du FT, ni le blocage de l’ensemble de la coagulation par la warfarine (un anticoagulant déplétant la vitamine K) n’ont permis de voir une différence dans le développement d’inhibiteurs chez ma souris166.
Intensité du traitement
Si certaines études montrent que l’âge au moment de la première injection de FVIII peut constituer un facteur de risque, cela reste à confirmer. En effet, la majorité des études sont rétrospectives et se fondent sur l’âge de la première détection d’inhibiteurs, qui peut différer du moment de leur apparition. De plus, d’autres études semblent contredire ces résultats167. En revanche, une étude s’attachant à l’intensité du premier traitement (dose
de FVIII) sur le développement d’inhibiteurs a été publiée en 2007. Cette étude rétrospective sur 366 PUPs a conclu notamment que le lien entre le bas âge lors de la première injection et le développement d’inhibiteurs décrit parfois, est probablement surtout dépendant de la dose de FVIII injecté168. Ces résultats confirment une observation
similaire faite chez des patients hémophiles A modérés169 et ont depuis été confirmés chez
des hémophiles A sévères170. Cela peut être relié à la « théorie du danger » développée
présence de signaux de danger, en plus de l’antigène. Cette théorie est supportée par l’observation que l’injection de protéines exogènes ne permet pas systématiquement le déclenchement d’une réponse immunitaire, et nécessite parfois un adjuvant, source de ces signaux de danger. Ainsi, les patients ayant le plus d’évènements hémorragiques aigus sont ceux qui reçoivent des fortes doses de FVIII thérapeutiques (en péri-opératoire par exemple) et sont également les plus susceptibles de développer de l’inflammation (suite à l’opération pour traiter le saignement aigu). Cette inflammation constituerait alors un signal de danger, propice au développement d’une réponse immunitaire anti-FVIII et donc d’inhibiteurs173. Cela est cohérent avec l’observation selon laquelle les patients atteints
d’hémophilie A sont ont un statut plus pro-inflammatoire que des sujets sains174.
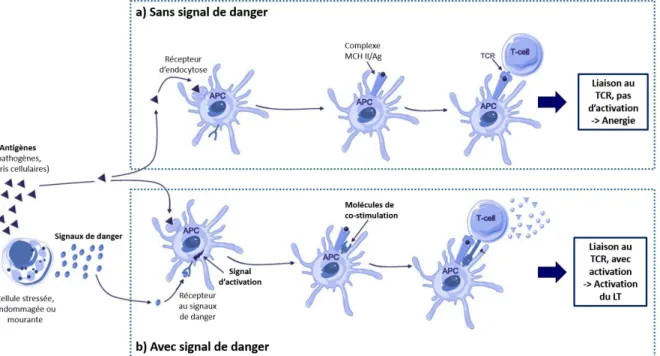
Figure 8. Représentation schématique de la théorie du danger. Lorsqu’un antigène (triangle) est reconnu par une cellule présentatrice d’antigène (APC) sans signal de danger (schéma du haut), la présentation des peptides au LT (T-cell) mène à son anergie. Lorsqu’un signal de danger (rond) est également fixé par l’APC en plus de l’antigène (schéma du bas), la présentation au LT mène à son activation. D’après Lövgren, Haemophilia, 2016.
Traitement prophylactique ou à la demande
D’autres études ont confirmé une augmentation du risque d’inhibiteurs de 2,5 fois en cas d’utilisation de fortes doses de FVIII (> 35 UI/kg)152.
Ces mêmes études ont également comparé l’utilisation de FVIII thérapeutique en prophylaxie ou à la demande. S’il a été montré que le traitement prophylactique diminue