A Photochemical Approach to Pyridopyrroloquinoline Derivatives as New Potential Anticancer Agents
6
0
0
Texte intégral
Figure
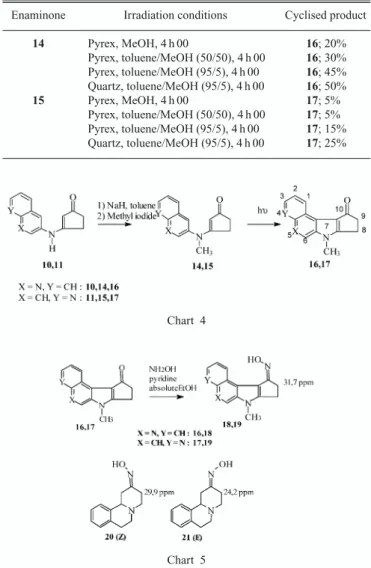
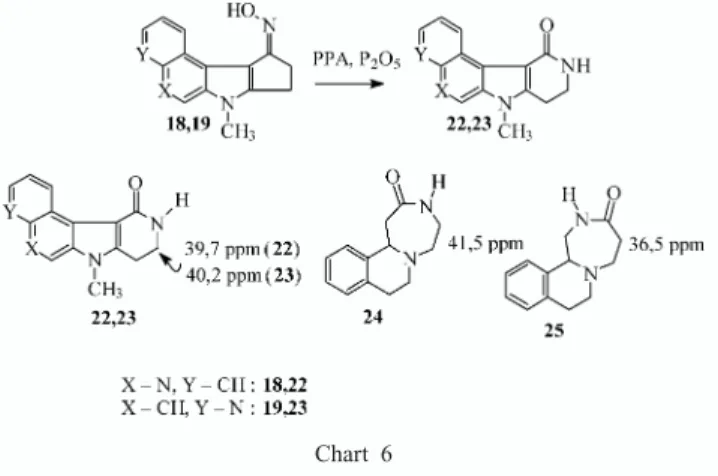
Documents relatifs