HAL Id: jpa-00210421
https://hal.archives-ouvertes.fr/jpa-00210421
Submitted on 1 Jan 1987
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
teaching and research institutions in France or
abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
recherche français ou étrangers, des laboratoires
publics ou privés.
Generalization of Heisenberg Hamiltonians to non
half-filled bands : a magneto-angular effective
Hamiltonian for boron clusters
A. Pellegatti, F. Marinelli, M. Roche, Daniel Maynau, Jean-Paul Malrieu
To cite this version:
A. Pellegatti, F. Marinelli, M. Roche, Daniel Maynau, Jean-Paul Malrieu. Generalization of
Heisen-berg Hamiltonians to non half-filled bands : a magneto-angular effective Hamiltonian for boron
clus-ters. Journal de Physique, 1987, 48 (1), pp.29-43. �10.1051/jphys:0198700480102900�. �jpa-00210421�
Generalization of
Heisenberg
Hamiltonians
to
nonhalf-filled bands :
amagneto-angular
effective Hamiltonian for boron
clusters
A.
Pellegatti,
F.Marinelli,
M. Roche(*),
D.Maynau
(+)
and J. P. Malrieu(+)
Centre de
Thermodynamique
et deMicrocalorimétrie,
(U.P.
7461 duC.N.R.S.),
26, rue du 141e R.I.A.,13003 Marseille, France
(+)
Laboratoire dePhysique
Quantique,
(U.A.
505 duC.N.R.S.),
Vniversité Paul Sabatier,118, route de Narbonne, 31062 Toulouse Cedex, France
(Requ
le 26 mars 1986,accepté
le 16septembre
1986)
Résumé. 2014
On propose un hamiltonien effectif pour l’étude des états neutres
d’agrégats
de Bore, engénéralisant
les hamiltoniens deHeisenberg
au cas d’une banderemplie
au 1/6e.L’espace
modèle estporté
par lesproduits
des fonctions fondamentales2P(s2p)
des atomes. L’hamiltonien effectifimplique,
outre des termesd’échange
despin
de typeHeisenberg,
desopérateurs d’échange
du momentangulaire
et desopérateurs changeant
à la fois lespin
et lemoment
angulaire
des atomes. Lalogique
de cet hamiltonien effectif est obtenue pardéveloppement
au3e ordre de la théorie des
perturbations
quasi dégénérées.
Enpratique
l’hamiltonien est obtenu directement par leséquations
de Bloch àpartir
de calculsprécis
des fonctions d’onde deB2.
La transférabilité de cet hamiltonien effectifa été testée sur le
système
B3
linéaire ; lapartie
basse du spectre est correctementreproduite.
Ce modèle sera utilisépour l’étude du
ferromagnétisme
possible
de chaînes linéaires de Bore. L’article discute le rôle del’hybridation
etsouligne
laportée
de cetteapproche, qui
étend la rationalisation par Anderson des hamiltoniens deHeisenberg
à dessystèmes
autres que les bandes 1/2remplies.
Abstract. 2014 An effective Hamiltonian has been
proposed
for thestudy
of the neutral states of boron clusters, i.e. for a 1/6th filled band. Thisgeneralization
ofHeisenberg
Hamiltonians rests upon the definition of a model spacespanned by
products
ofground
state2P(s2p)
atomic wave-functions. Theresulting
effective Hamiltonian involves, besidesHeisenberg-type
effectivespin exchange
terms,angular-momentum
exchange
operators andspin-and-angular-momentum exchange
operators. Thelogic
of this effective Hamiltonian has been derivedthrough
3d orderquasi-degenerate perturbation
theory.
Inpractice
the effective interactions are obtained from accurate MO-CIlarge
basis set calculations onB2 through
the use of aspectral
definition ofHeff.
Thetransferability
has been tested on the linearB3
system,by comparing
theHeff
predictions
with accurate MO-CI results. Thetransferability
proves to besatisfactory
for the lower part of the spectrum. The model will be used for the treatment of themagnetic
character of(possibly high spin) Bn
linear chains. The role ofhybridization
processes has been discussed and thepossible
extension of thisapproach,
derived from Anderson’s rationalization ofHeisenberg Hamiltonians,
has been outlined. ClassificationPhysics
Abstracts 31.10 -31.20D - 31.20R - 75.10
1. Introduction.
The theoretical foundations of
Heisenberg
Hamil-tonians[1]
are known to be based on thetheory
of effective Hamiltonians and on the use ofquasi-degen-erate
perturbation theory
[2].
These effective Hamil-tonians are easy to derive forsystems
involving
oneelectron in one Atomic Orbital
(AO)
on each centre[2],
and(although
lessevidently
[3])
forsystems
(*)
Permanent address :Ddpartement
de Chimie, Univer-site de Provence, Place VictorHugo,
13331 Marseille Cedex 3, France.involving
p electrons in p AO’s per centre. In bothcases the
systems
involve a half-filled-band(the
other bandsbeing
full orempty).
Besides theirtypical
applications
for Mott insulators andmagnetic
problems
in coordinationchemistry,
theHeisenberg
Hamil-tonians haveproved
to besurprinsingly
efficient tools forproblems
where the electronics delocalization is far frombeing
small,
namely
inorganic chemistry
for thestudy
ofconjugated
molecules[4]
and even for alkaliatom clusters or solids
[5].
The basic idea of the
present
work is toattempt
anextension of this
approach
to non-half filledbands,
forinstance to 1/6-filled
bands,
as will occur ins2p
atoms(B
or Alclusters).
The motivation and thestrategy
aredeveloped
in section2 ;
the choice of the model space,spanned by
the Valence Bond(VB)
determinants built fromground
state2p
( s2p )
atomicwave-functions,
isexplained
and discussed. Theferromagnetic
character of theB2
ground
statesuggests
apossible
ferromagnetic
ordering
of the linearBn
chains.Section 3 derives the formal structure of the effective
Hamiltonian from the
application
of thequasi-degener-ate
perturbation
theory
(to
thirdorder)
to theB2
problem.
It is shownthat,
besidesangular dependant
repulsion
terms and other first order terms, the effective Hamiltonianpresents
alarge
variety
ofoperators,
of second-and third-orderorigin.
Theseoperators
act onlocal
spins only, angular
momentaonly
or on bothspin
andangular
momenta, andimply
2 AO’sonly
(of
either the same ororthogonal
subsystem)
or 4 AO’s. The effective Hamiltonian for thisproblem
is thus amag-neto-angular
effective Hamiltonian since it acts on boththe local
(i.e. atomic) spin
andangular
momenta. Its formal structure isdeveloped
insecond-quantization
formalism. From accurate molecular orbitalconfigura-tion interaction
(MO-CI)
andorthogonal
V.B. calcula-tions onB2
numerical effective Hamiltonians arepro-posed, refering
to a direct(non-perturbative)
definitionof the effective
Hamiltonian,
analogous
to those of Bloch[6]
and Des Cloiseaux[7].
In a fourth section the
transferability
of theseeffec-tive Hamiltonians to heavier
systems
is tested on thelinear
B3
problem.
For this molecule accurate MO-CIcalculations have been
performed assessing
the exact stateordering.
It appears that in thissystem
all the lowest neutral statesessentially
keep
thes2p
character of the atoms. The effectiveHamiltonian,
extractedfrom the relevant lowest
eigenstates
ofB2, gives
surprisingly
accurateenergies
for all the lowest states ofB3
of variousspin multiplicities
andsymmetries.
Thisimplicit
treatment of thehybridization problem
isdiscussed and a further
generalization
of theapproach
to the simultaneous treatment of different bands is
suggested. Applications
tolonger
linear chains isplan-ned for the near future.
2. Situation of the
problem.
2.1 HALF-FILLED BANDS : THE HEISENBERG HAMIL-TONIANS. - For
a
system
involving n
electrons and nactive
AO’s,
theprocedure
to obtainHeisenberg
Hamiltonians consists first in the definition of a model space ; this model space is
spanned
by
the Valence Bondsingle
determinants which occupy each of the n active AO’s of thesystem
once andonly
once. Then all the determinants of the model space have the samespace
part ;
they only
differby
thespin
distribution,
and the model space is
composed
ofC :/2
determinants.These determinants are all neutral in the sense of V.B.
theory,
andthey
are thedeterminants
of lowest energyin the V.B. matrix
(at
least innon-polar
well-definedproblems).
The secondstep
of theprocedure
consists inapplying
theQDPT
[8]
to this model space, i.e. to constructperturbatively
the effectiveenergies
and effectivecoupling
of the neutraldeterminants
under the influence of the ionic determinants. For atwo-electron,
two-AO(a
andb),
two-centresystem,
Ander-son shows that the effective
exchange
between
the twoneutral
determinants
I
ab
I and
lab
l
iswhere
Kab
is the direct bicentricexchange, Fab
thehopping
integral
between the two centres and AE is the« transition »
energy from the neutral to the ionicI aal [ or
lbbl
I
determinants. TheHeisenberg
Hamil-tonian treats the electronic delocalization
(i.e.
the mixture between neutral and ionicdeterminants)
aseffective
spin
operators
between the neutral deter-minants.This
straight
forwardapproach
is notdirectly
generalizable
to thegeneral
half-filled bandproblems
(p
electrons in p AO’s percentre),
if one uses thewell-known Bloch
[6]
or Des Cloizeaux[7]
definitions of theeffective
Hamiltonian,
as shownby Maynau et
al.[3]
recently ;
new definitions of the effective Hamiltonianare to be
proposed
to obtain agood
transferability
ofthe effective
couplings
from the two-centreproblem
tolarger
systems.
Nevertheless thegeneral
foundations of theHeisenberg
Hamiltonians for half-filled bandprob-lems are clear.
The
Heisenberg
Hamiltonians haverecently
receivednew
applications,
far from thetypical
field of Mottinsulators.
They
have beenapplied
withsurprising
success to the
7r-systems
ofconjugated
molecules inorganic chemistry [4].
Aprecise
MO-CI calculation onethylene
makespossible
the accurate extraction of twooperators
(a spin-dependant
opotential
+ 7rrepulsion
and an effectiveexchange)
which are functions of theinteratomic distances. This
Heisenberg
Hamiltonianpredicts
veryaccurately
thegeometries,
isomerizationenergies,
rotationalbarriers,
lowest excited statesener-gies
and conformations ofconjugated
moleculesinvolv-ing
up to 10 carbon atoms[4].
A similar
attempt
has beenperformed
on anisoelec-tronic
problem, namely
the alkali atom clusters. AHeisenberg
Hamiltonian has been extracted fromaccu-rate MO-CI
potential
curves ofNa2 ;
it proves togive
realisticenergies
of small clusters and the cohesiveenergy of the bulk
[5].
The
application
of ab initio extracted efficientHeisenberg-type
Hamiltonian for thestudy
of metal clusters may appear as atempting challenge
in view ofthe
difficulty
of usual ab initio MO-CItechniques
for the treatment of such versatilesystems,
presenting
certainly
severalnearly degenerate optimal
conforma-tions withpossibly
differentspin multiplicities.
2.2. NON HALF-FILLED BAND : TOWARDS NEW TYPES
OF EFFECTIVE HAMILTONIANS. - The basic purpose
of the
present
paper is to establish the form of aneffective
Hamiltonian, inspired by
thestrategy
leading
to the
Heisenberg
Hamiltonians butdealing
with nonhalf-filled bands. The fundamental idea is
that,
as inAnderson’s
legitimization
of theHeisenberg
Hamil-tonian,
one may:i)
choose as model space the set of neutraldetermin-ants with maximum
spread
of the electrons in the concernedband,
ii)
apply
thequasi
degenerate
perturbation
theory,
or an alternative variational formalism
[9],
to build the effective Hamiltonianspanned
by
this model space, to treat the effect of ionicconfigurations
on the neutralsubspace.
As a model
problem
we consider a one-sixth(1/6)-filled
band,
i. e. a cluster of B(or Al)
atoms in theirs2p (2P)
ground
state.The s band issupposed
to be filled up, and thehybridization
will be considered as aperturbation only.
We choose the model spaceSo
asbuilt of all the determinants in which each atom A bears one
(and
only
one)
p-electron
in a PXIPY
or p,AO. The
projector Po
on the model space is written as :where
Then it is clear that the model space determinants do not have the same
space-part ;
for each atom one hastwo
degrees
of freedomnamely
theorientation x, y
or zof the
occupied
AO and itsspin.
The size of theeffective matrix for a 2 n-atom
problem
is32n
C2n,
larger
than for atypical
Heisenberg
Hamiltonian,
buttremendously
smaller than the full V.B.-matrix size. Thelogic
of the effective Hamiltonian is of coursedifferent from that of a
Heisenberg
Hamiltonian sinceit deals with both the local
spin
andangular
momentum.One may
expect
that the effectiveoperators
will beboth bicentric
spin exchange
operators
andI-exchange
operators,
as will be confirmed in the next section. Thelogical
structure of the effective Hamiltonian has to be established fromanalytic perturbative developments
using
thequasi degenerate perturbation theory
Qn atwo centre
problem
in thespirit
of the Anderson-Brandowapproach
toHeisenberg
Hamiltonians.Two
types
ofproblems
may arisealong
thispath :
-
non-convergence of the
quasi
degenerate
pertur-bation
expression, especially
at shortdistances ;
- mixing
withhybridized
structures in which thesp2
excited atomicconfiguration
becomes dominant. Theseconfigurations
may appear as intruder states inJOURNAL DE PHYSIQUE.-T. 48, N° 1, JANVIER 1987
the
QDPT
expansion,
questionning
thevalidity
of the choise of the model space. Thisproblem
will bebriefly
discussed in the
present
paper but theperturbative
difficulties will be avoidedby
aspectral
definition ofthe effective Hamiltonian. The
generalization
of thisapproach
to the simultaneous treatment of severalbands is out of the scope of the
present
work. 2.3 INTEREST IN B CLUSTERS. - At least atlong
distances,
the reduction of the model space to thedeterminants built from the
s 2p (2p)
ground
state of the atoms isvalid ;
the lowestSp2 (2p)
atomic state is 3.5 eV above theground
state, and thecorresponding
Sp2 (2 D )
is 6 eV above theground
state. Theground
state of
B2
is of3-v;
symmetry,
isessentially
built fromA ( S2px) x B ( S2py )
atomicconfigurations,
and isferromagnetic :
This
point
isinteresting
since itsuggest
that :i)
thes2p
configuration
may dominate theground
state of at least linear B clusters.ii)
the linear B chain may be ahigh
spin compound
since it may be viewed as dominated
by
aspin-angular
arrangement
i.e.presenting
a molecularmagnet.
Be-sides the formalproblem
treatedhere,
thepresent
work may then be ofpractical
interest.3. Structure of the
B2
effective Hamiltonian.3.1 LOWER PART OF THE SPECTRUM FROM A MO-CI
APPROACH. - The number of papers related to be
B2
molecule is rather limited.Among
thoseconcerning
the order of thelow-lying
states, let us note first the calculations ofPadgett
andGriffing
[10]
at the SCFlevel and of Bender and Davidson
[11]
at the CI level. In thelast,
the CI was notsufficiently
extended so thatthe
5E u
isassigned
to theground
state in both papers,instead of the true
3 V9
state[12].
At the sametime,
Bader,
Henneker and Cade[13]
focused their attentionon the
charge
distributions and the chemicalbinding.
More
recently Dupuis
and Liu[14] performed
a moreextended CI calculation and found a
good
agreement
between their results and
Douglas
andHerzberg’s
experimental interpretation
of thesymmetry
of theground
state( 3 Ii)
and of the first303A3 :> 3 Ii
transition. The
51 u
state was found 0.006 a.u.higher
than the
ground
state. Becke[15] using
a numericalHartree-Fock-Slater method studied
only
theground
state303A3g.
He found a dissociation energy of 3.8eV,
slightly
larger
than theexperimental
valueFig.
1. - MO-CI[xy] -symmetry
potential
curves ofB2.
Fig.
2. - MO-CI[xx,
yy,zz]
-symmetrypotential
curves ofB2. L1
states,degenerate
with those of the[xy ]
-symmetry have been removed from thefigure.
( 3.1
± 0.2 eV[16] ) .
All these calculations found aninteratomic distance close to the
experimental
value 3.005 a.u.[17].
The most accurate results are those ofDupuis
and Liu butunfortunately
they only
concernthe X
3 Xi ’ a 5X;;
and the two first303A3u;
states. As will be seen in nextsections,
a series ofpotential
curves of
B2
arerequired
for the construction of thedesired effective Hamiltonians.
By
the way, it is alsointeresting
to test thevalidity
of our MO-CI calculationsfor this
simple
case and inparticular concerning
the small differenceseparating
the3 Xi
and5 X;;
states. At the SCF level we used the Pshondo program[18],
anextansion of Hondo
[19].
From the basis evaluated for the B atom[20]
we extracted atriple-ts
(contracted [3,
1, 1]),
adouble- 03B6p (contracted [3, 1])
and onesingle- 03B6d
basis set. The
d-exponent
parameter
was chosenequal
to 0.5. Correlation effects have been introducted
by
means of the CIPSI
algorithm
[21].
In thismethod,
amulticonfigurational
zero-order wave function is firstobtained from a variational
configuration
interaction(CI)
calculation in a restrictedsubspace
built up from the mostimportant
determinants for the statesbeing
studied ;
then this wave function isperturbed
up to second order in energy.The
319
state isclearly
found to be theground
state. Thespectroscopic
characteristics(re
= 3.05 a.u.,D,
= 3.10eV)
are ingood
agreement
withexperiment
(re
= 3.005 a.u.,D,
= 3.10eV).
As alsopredicted by
Dupuis
andLiu,
the5 X;;
state is obtained at 0.01 a.u.above the
ground
state, with aslightly
smallerequilib-Fig.
3. - MO-CIrium distance
( re
= 3.00a.u..
One mustimmediately
notice that this503A3u;
state isnecessarily
spanned
by
BSp2 (2 D)
x Bs2p
(2p )V.B.
structures, and it isneces-sarily
outside of our model space(cf.
Eq.
(1)).
The relevant
potential
curves ofB2
arepictured
infigures
1-3. Curvescorresponding
to various irreduct-iblerepresentations
of theD 00 h point
group have beengathered
into three reduciblerepresentations,
which may be reached from the use of px p, and pZ AO’s(z
being
the internuclearaxis) ;
thispresentation
isespe-cially
convenient for theforthcoming
construction of the effective Hamiltonians. Thefigures only
report
thepotential
curves of the stateshaving
significant
compo-nents in the model space(with
theexception
of the above mentioned503A3u-
state, which appears as a dottedline).
The concerned states arealways
the lower ones of theirsymmetry,
with threeexceptions only :
thehighest
in eachsymmetry.
Other states, of redundant(spin
orspace)
symmetry
and/or of irrelevant V.B. characterwere found between the two
reported
303A3u ,
30394u
and1 _Vu,
3 llg
and1 llg
states. Inparticular,
one noticesirregular shapes
of the3IIg
and1 llg
potential
curves,which result from an avoided
crossing
with upperconfigurations.
Since thecorresponding eigenstates
kept,
at short interatomicdistances,
anon-negligible
(although non-dominant)
component
in the model V.B. space, we decided to use these states in thedefinition of the
« target »
stablesubspace
defining
Heff.
This may be called an adiabatic definition of thetarget
space S(see
below for a more detaileddis-cussion).
3.2 DEFINITION OF THE EFFECTIVE HAMILTONIAN. 3.2.1 Model space, size and
symmetry
partitioning.
-According
to the basic choice of our treatment, the model space forB2
reduces to the neutralA ( S2p )x B ( S2p )V.B.
determinants(cf.
Eq.
(1)).
where
PA,i
is aspin
orbitalPx,
y or z on the atom A. The model spacesplits
intoSz = ± 1, SZ = 0
indepen-dent
subspaces,
the last onebeing
thelargest
one andcontaining
all the relevant information.Equations (1)
and(2) implicity
assume the use oforthogonal
valenceAO’s. This may be achieved
by
apreliminary
S- 1/2
transformation,
but,
as noticed elsewhere[4,
5],
theprecise
specification
of this basis is not necessary. Thisorthogonality problem,
connected withtransferability
tolarger
systems,
will be discussed in thesubsequent
section. This model space may besplit
into threedifferent
symmetries
[xa Yb, y a xb
andxa
Zb’ ZaXb]
(and
itsdegenerate
[ya
Zbl ZaYb
counterpart)
where the atoms arede-noted a and b for convenience. The size of the effective matrices will be
6,
4 and 4respectively for Sz
=0,
and3, 2, 2,
forSz
= ± 1. The u, gsymmetry
will beexploited
later on.Once the model space is defined the effective
Hamil-tonian may be obtained
by
two different routes :- the
perturbative approach, using quasidegenerate
perturbation theory
in one of itsversions ;
- the direct use of the Bloch or Des Cloiseaux
equations,
since in thatprecise problem,
almost exactenergies
and wave functions are available from theMO-CI
preliminary
calculations. The latter solution isequivalent
to an infinite order summation of theperturbative
expansion,
and isby
far moreprecise,
butthe low-order
perturbative
approach gives
the main structure of the effective Hamiltonian and thephysical
origin
of the effective interactions.3.2.2 Structure
of
theeffective
Hamiltonianfrom
third orderperturbative expansion.
- Some details about thegeneral
structure weregiven
inprevious
papers[3-5].
Let us recall that theperturbation
operator
is definedby :
with
where
So
is the model space. In anEpstein-Nesbet
definition
[22]
ofHo,
the Voperator
ispurely
ex-tradiagonal. Up
to second order the effective Hamil-tonian matrix elements take the form:and to third order
where
a, (3
may be ionic orhybridized
neutral deter-minants.Since the ionic determinants are known to
play
the dominant role in theinterpretation
of the effectiveantiferromagnetic coupling
in half-filledbands,
we shall concentrate our attention on their effect in ourdetermin-ants to those which
keep s2
electronpairs
on eachatom,
the V.B. matrix may beschematically pictured
as infigure
4,
for the[xx,
yy,zz]
symmetry,
which is thelargest
and mostcomplex
one. The zeroth orderenergies
of the neutral determinants arerepulsive,
but to alarger
extent for za zbspatial
parts
(Rzz:> Rxx ) ;
the same differences appear among ionic states. Thelargest
extradiagonal
elements arehopping integrals
Fcoupling
theneutral xa xb (resp. ya yb
or zaTb)
and ionicxa xa (resp. Ya 37a
orza ia)
determinants. One should also notice the occurrence ofnon-negligible
monocen-tric
exchange integrals K
between ionic determinants.The
hxy
(and
hxz)
integrals
represent
electrostatic in-teractions between the atomic xy(or xz) quadrupoles ;
the otherintegrals
involve bicentricoverlap
distribu-tions and are much smaller.
To first
order,
the differentsymmetries
involve 6parameters
The
identity
ofthe dxy
anddxz_y2
solutions at first order leads to thefollowing
relationStarting
from the neutral xaxb
determinant one maygo to an ionic xa
xa or xb xb
determinant. From it onemay either move back to
the xa xb
original
determinant(and
obtain adiagonal
second ordercorrection)
orreach the
x b , 7
determinant ;
the effectivecoupling
is then an interatomicexchange,
shown in thefollowing
scheme where * and 0 stand for a andp
spins
respectively.
This back-and-forth second order process in the
’IT x
subsystem
repeats
the well-known Anderson’s mechanism ofantiferromagnetic
coupling.
One maywrite
diagonal - gxx IXa Xb) (xa xb l
andoff-diagonal
- gxxlxaXb) (xbxal
operators,
withg = gxx
=2
F2/AExx,
whichgive
theamplitude
of thenegative
effective
exchange integral
in the ’IT xsystem.
Analogous
contributions occur in the
’IT y (gyy = gl)
andu (gzz = gí)
systems.
A second
type
ofcoupling
occurs at thirdorder,
pictured
as followswhich
couples xa xb and ya yb
through
(the
factor 2 reflects thepossible
travelthrough
A+ A- and A- B+
structures).
Thisoperator
changes
theangular
part
of thewave-function,
and therefore cannot enter aHeisenberg
Hamiltonian.Analogous
operators
occur between x and z or y and zspatial
parts,
with a differentamplitude
exz =Eyz.
Table Isummarizes the
expected
structure of the effectiveFig.
4. - Structure of the V.B. matrixspanned
by
the nonhybridized, Sz
=0, determinants
in the[xx,
yy,zz]
-symmetry. Bicentricexchange integrals
are not taken into account. Identities between matrixelements,
resulting
from thedegeneracy
of 7rHamiltonian
in the[xx,
yy,zz]
symmetry
(Sz
= 0and 1).
In the
[xy ]
symmetry,
analogous
processes takeplace
leading
to second orderdiagonal
andoff-diagonal
corrections,
with for
example
thecorresponding off-diagonal
operator
where one may notice that
AExy =
AExx -
2 Kxy,
since one does not gothrough
an ionic determinant with 2electrons in the same
AO,
as occurred in the Andersontypical
problem
and in the[xx, yy, zz]
symmetry.
A third order contribution also appearsthrough
processes such asleading
tonon-Heisenberg
operators
Table I
gives
the structure of the third order effective Hamiltonian in the[xy]
symmetry
forSz
= 0 andSz
= 1. Theidentity
ofthe SZ
= 0and Sz
= 1triplet
eigenvalues requires
thatThis relation is
easily
understood from the difference inthe energy denominators
so that
Similar derivations are
posible
for the[xz]
(and
[yz] )
symmetry
and are also shown in table I. However oneshould notice that :
i)
thediagonal
second order corrections arewhile the
off-diagonal
second-order correction obtainedfrom the
operator
is of different
amplitude
than thediagonal
correctionand
ii)
theFzz hopping
integral
ispositive,
resulting
inapparent
changes
insign,
also active inthe £3
thirdorder terms.
The
identity
ofthe 3
Sz
=0)
and 3
Sz = 1)
solu-tions for the[ xz ]
symmetry
leads to thefollowing
relations
analogous
toequation
(3).
3.2.3
Physical typology of
theeffective
operators.
-One may summarize the various
types
ofoperators
according
to several criteria :-
diagonal/non-diagonal
character ;
- order ofperturbation
concerned ;
- number of AO’sinvolved ;
- actionon the local
spin
andangular
momenta. On thediagonal
one first haspurely
( la, lb)
depen-dent terms R which are
independent
of thespin.
One alsohas - g
stabilization second order contributions.The terms
only
involve two AO’s of the samesubsystem,
andappear when these two AO’s bear electrons of different
spins.
The -
g2/xa Y7b) 1xa 3Tbi I - g31xa Zb) xa zb l
termsimply
two AO’sbelonging
to twoorthogonal
subsys-tems.
The
operators
appear inthe Sz
= 1(or -1)
subspaces. They
involve two AO’s of two
orthogonal
subsystems bearing
parallel spins.
Off the
diagonal
one findstypical
second orderspin
exchange integrals involving
only
two AO’s of the samesubsystem,
as in allHeisenberg
Hamiltoniansand
analogous
operators
foryy ( - gl)
andzz ( - g1 )
determinants.Other second order
operators
of thetype
Table II. -
Characters
of
the variousoff-diagonal effective
operators.momentum. These
operators
involve four AO’sbelong-ing
to twoorthogonal subsystems.
The second orderoff-diagonal
operators
change
theangular
momenta of the atoms withoutchanging
theirspins.
One should notice that due to theidentity
of the ’IT x and’IT y systems ii2
= 92and g
=g2.
The third order
operators
change
thespins
of twospin-orbitals
of differentspins
belonging
to twoorthogonal
subsystems,
withoutacting
on the space
part.
The third-order
operators
change
theangular
momentum on the atom withoutchanging
the localspin ; they
involve four atomic orbitalsbelonging
to twoorthogonal
subsystems.
Finally
the third-orderoperators
change
both theangular
andspin
momenta on theatom,
involving
therefore four AO’s on twoorthogonal
subsystems.
Thistypology
isclearly
pictured
intable
II. 3.3 AB INITIO DERIVATION OF THE EFFECTIVE HAMILTONIANS FROM THE BLOCH EQUATION. -Since the effective Hamiltonian satisfies the Blochequation
for n rootswith
a
spectral
definition ofHeft
isgiven
by
where
Po 03C81
is thebiorthogonal
vector associated with theprojection Po
03C8i
of theeigenstate
03C8i
of H intoSo.
If the exact
spectrum
of H and itseigenvectors
areknown,
a selection of neigenvalues
andeigenvectors
03C8i
definesthe stable
subspace,
which we call «target
space », andHeff
isentirely
definedby
P 0’ P s
and the exacteigenenergies,
without any reference to apossibly
divergent
perturbation
expansion.
If one uses the waveoperator
n,
then :Since for the
B2
molecule,
accurateenergies
andwave functions are available from the MO-CI
cal-culations,
it istempting
to useequation (5)
to obtaindirectly
theHeff
matrix,
in apurely
numerical form : inthis section the
target
space is defined asspanned by
the
eigenvectors
of the correctsymmetry,
lowest energy andhaving significant
components
on the model space.Another definition of the
target
spacemight
pick
up theeigenvectors having
thelargest
components
onto themodel space. From
symmetry
considerations,
thefol-lowing correspondence
between modelsubspaces
andThe A states obtained from
[xx, yy, zz ]
and[xy ]
model
subspaces
are of coursedegenerate.
All the vectors which are the
only
ones of theirsymmetry
in the concerned modelsubspace
have theircomponents
in the model spaceentirely
definedby
symmetry.
This means that thecomponents
ofP 0 f/J
i areimmediately
obtained. For instance for3-v9
onenecessarily
has +--+components
on thexa Yb ,
Iyb xa ,
IYa
xb
y and
xbYa
model
determinants. Theonly
exception
concerns thepairs
ofI!;
and303A3u+
statesbelonging
to the[xx,
yy,zz ]
symmetry.
Thedegree
offreedom concerns of course the ratio of
Xa 3Fb I ( =
IYa Yb I)
overza
zb
I
components.
In
principle
theprojected
eigenvectors
of the samesymmetry
are notorthogonal
This results in a non-Hermitian structure of the Bloch effective Hamiltonian. For the sake of
simplicity,
wepreferred
to use Hermitian structure. Onepossible
solution refers to an
S-ll2
transformation of theprojec-tions,
and is known as the des Cloizeaux[7]
effectiveHamiltonian definition. This solution
symmetrically
distortsprojections ;
previous
work[3]
onHeisenberg
Hamiltonians for a half-filled band with 2 e-
/2
AO percentre has shown that this distortion may result in
important
deviations from theexpected
structure anddifficulties in the transfer of effective
operators
tolarger
systems.
A better solution consists of aSchmidt-type
orthogonalization keeping
the lowestprojected
eigenvector unchanged,
andorthogonalizing
the secondone
(of
higher
energy)
to the first one. This solution has beenapplied
forlz+
and303A3
symmetries.
The ratios of
I xa
3Eb
l (
=ya
j7b
overza
zb
I
pro-jected
components
aregiven
in tableIII,
otherpro-jected eigenvectors
on the model spacebeing trivially
obtained
by
symmetry.
In thistable,
the first state of eachsymmetry
corresponds
to the lowest in energy.The
changes
of the order ofmagnitude
(for
103A3g;
states)
and of thesigns
(for
303A3u:
states),
between the two statesof each
symmetry,
occurring
between 3.3 and 4.5 a.u.,both
correspond
to avoidedcrossings.
For 3.3 a.u., thetwo ratios smaller than 1.0 found for
103A3g+
states do notmean that the states are both «
z2 dominant
» states,since the
[xx] -component
has to be counted twicexx
I and
[ yy I
determinants).
The first’,V+
state is axx
,I YY I ] -dominant
state. Thecorresponding
ef-fective Hamiltonians are
given
in tableIV,
where theenergy of the two
separated
atoms(5.18 a.u.)
has been taken as zero of energy.3.4 COMPARISON BETWEEN THE PERTURBATIVELY
DERIVED AND NUMERICALLY OBTAINED EFFECTIVE
HAMILTONIANS. - For the
diagonal
R «repulsive
»matrix elements one should notice
(cf.
Sz
=1,
Exx, yy . zz ]
symmetry)
thatRzz ( = R[)
is the mostrepulsive,
asexpected.
Thelarge
off-diagonal
matrixelements
correspond
to the effectiveexchanges
g. Sincethe
overlap
(and
hopping
integrals)
are oflarger
amplitude
in theu-system
than in the 1T ones, oneshould have
The numerical values
actually
show thelarge
9 uu value(0.115 a.u.)
at r = 3 a.u., but the three otherintegrals
are of the same order of
magnitude
(-
0.03 to 0.02a.u.).
One should notforget
that our third-orderperturbative
development
did not involvehybridized
configurations,
and deviations arelogically
expected.
Nevertheless the numerical effective Hamiltonian
keeps
the
predicted
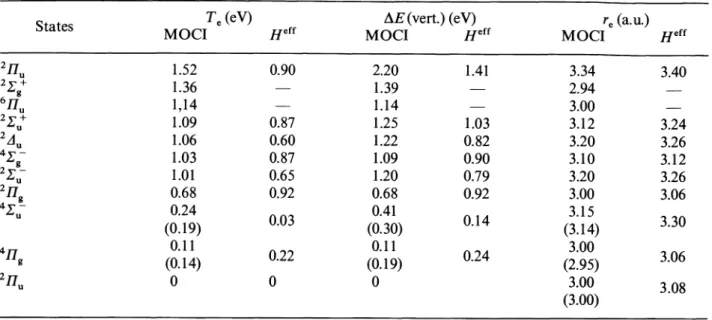
structure.Table- III. - Ratio
of the projected
components
of
xa
Xb
[ over
za
ib
I
for
the states in the[xx,
yy,zz]- symmetry,
Table IV. - « Exact» numerical
Heff
matrix elements at various B-B distances. Deviationsfrom
thetheoretical
third order structure are
clearly
outlinedby
a directcomparison
to the.secondcolumn.
The effective Hamiltonians have been calculated for five interatomic distances. All matrix elements
R,
g, E decreaseexponentially
with the interatomicdistance,
since
they
involve interatomicoverlap
distributions. Thequadrupole-quadrupole
interactions h decrease asr-
8.
Analytic
fittings
of the matrix elements are easyand may be used for
geometry
optimizations
inlarger
clusters.4.
Transferability
to the linearB3
system.
Transferability
of the effective Hamiltonian has been tested on thesimplest larger problem, namely
theB3
linearsystem
keeping D 00 h
symmetry.
4.1
QUALITATIVE
CONSIDERATIONS. - Close to theequilibrium
geometry,
the lowest states ofB2
are, inincreasing
order :303A3g and 1 L1g
( [xy ]-symmetry
L-, )
and
3IIu
([ xz ] -symmetry: L-).
Starting
from two Binteractions to form the linear
B3
molecule,
we maypredict
tofind,
as lowest states forB3,
a structureresulting
from thejunction
of two such[xy ]-structures
or
one[xy ] -
and one[xz ] -structure : , ,
Moreover,
as also outlined in theintroduction,
thedistribution
corresponding
to allspins parallel,
i. e. aquartet
symmetry
in that case, should be low in energy.4.2 AB INITIO MO-CI CALCULATIONS. - Accurate MO-CI calculations have been
performed
onB3.
Thesymmetry
of theB3 ground
state wasinvestigated
first.Keeping
in mind that d-functions were of fundamentalimportance
in the determination of theright
symmetry
for theground
state ofB2,
we used the sametriple-Cs,
double-( p
andsingle-Cd
valence basis set as forB2.
According
to the CIPSIalgorithm,
agreat
part
of thevalence correlation energy has been treated
variational-ly
and theremaining
part
perturbatively.
Figure
5 shows the three lowest states close to theequilibrium
internuclear distance which is found of the same order as that of
B2.
The21Iu
state is theground
state. Thequartets
4IIg
and4 _Vu-
suggested
by
ourqualitative
Fig.
5. -MO-CI
potential
curves for the three lowest statesof B3.
ground
state, the4IIg
statebeing
slightly
more stablethan the
4_V u -state.
In a second
step,
in order tolighten
thecalculations,
we eliminated the d-functions from the basis set.
Figures
6a and 7a show the lowest states for the threesubsymmetries.
These threesubsymmetries
are denotedhere:
[xxx]
(which
alsoincludes
I XYY l and
I xzz
l
determinants and the
degenerate
ones),
[zzz]
(with
also
[ xxz
I
and yyzdeterminants)
and[xyz].
The uand g characters are then
assigned
appropriatly
to each MO in MOCI calculations while a, b and csubscripts
characterize the atoms to each A.O. in the V.B.
formalism. The three lowest states are still found to be :
2IIu,
4 IIg
and4,X.-
in the same order.4.3 COMPARISON BETWEEN AB INITIO AND
EFFEC-TIVE HAMILTONIAN RESULTS FOR
B3.
- Malrieu etal.
[5]
discussed in detail theproblem
of theorthogonal-ity
between A.O.’s. Theorthogonalized
A.O.’s. for atwo-atom
system
and those for a three-atomsystem
arenot the same. We must
keep
in mind that this could affect thetransferability
to some extent. Their calcula-tions on alkali metals also have shownthat,
asexpected,
three-body
increments5 gab( c )
aresmall,
at least in linear chains.The
B3
effective Hamiltoniansplits
into three blocks of dimensions18,
21 and 21 for the[xyz], [xxx]
and[zzz]
pseudo-symmetries respectively.
Thelong-dis-tance
(A-C)-interactions
have been checked to havenegligible
effects. Theground
state iscorrectly
pre-dicted to be of2H .
symmetry
by
ourmodel,
and theFig.
6a. - MO-CI[xxx] -symmetry
lowestpotential
curvesof B3.
Fig.
6b. -Heff
[xxx]
-symmetry lowestpotential
curves ofFig.
7a. - MO-CI[xyz] -
and[zzz]-symmetry
lowestpotential
curves ofB3.
Fig.
7b. -Heff
[xyz ] -
and[zzz ]
-symmetry lowestpotential
curves of
B3.
equilibrium
distance is correct(3.08
a.u. to becom-pared
to 3.01 in the ab initiocalculations).
From a V.B.point
of view it may beanalysed
as a resonancebetween
The two upper states are very close in energy ;
they
arequartets
of4ng
and403A3u-
symmetries.
The best MO-CIcalculation
predicts
them atTe
= 0.14 eV and 0.19 eVabove the
ground
stateminimum,
while the effectiveHamiltonian
predicts
0.22 and 0.03 eVrespectively:
this inversion
corresponds
to an error of = 0.2eV,
which is not too bad.
Deleting
the d A.O.’s in the MO-CI calculation leads toTe
= 0.11 and 0.24 eVrespect-ively
for4IIg
and4!u.
Theimportant
feature is the differential effect on the interatomic distances(cf.
Figs.
6-7 and TableV)
which arecorrectly
reproduced
by
the effective Hamiltonian. The V.B. content of these wave functions may bepictured
asA direct
comparison
between thepotential
curves ofupper states may be obtained
by comparing figures
6a and 6b for the states of II and 0symmetries,
andfigures
7a and7b
for the states of-V-, d
and11
symmetries.
The results are also summarized in table V. There is
a
significant
gap(= 0.5 eV)
between the three loweststates and a group of five states which lie in a narrow
energy range of 0.4 eV. These states are of
2 llg,
2_Vu,
4 _V g
2 Llu
and2.I;
character,
in order ofincreasing
energy in the MO-CI calculation. The effec-tive Hamiltonianonly
permutes
theposition
of theZIIg
and2 Llu
states in this group. Thelargest
equilibrium
distances concern the2.I¡;
and2 Llu
states in bothapproaches.
The two next states are
6Hu
and2.I:
in the MO-CIg
calculation. The former cannot be reached from a
three-open-shell
model space and the second one isessentially
hybridized ;
from thatregion important
discrepancies
begin
to appear between the MO-CI andeffective Hamiltonian
spectra.
5. Conclusion.
The
major conceptual
interest of thepresent
derivationconcerns the
logical possibility
and cost of an extensionof the
Heisenberg
Hamiltonian to a non half-filled bandproblem.
The basicphilosophy
isagain
the choice of aTable V. - Lowest
spectroscopic
statesof
B3.
The values inparentheses
are relative to thelargest
basis set.and the use of
quasi
degenerate perturbation theory
orspectral
definition of the effective Hamiltonian. Theuse of the
perturbative expansion
has theadvantage
ofputting
inplace
thelogical
structure ofHeff,
showing
which are theleading
terms and theirphysical
origin.
The use of the
spectral
definition ofHeff permits
avoidance of
divergence problems
of theperturbative
expansion
and introduceshigh-quality (infinite order)
information.The effective
operators
involve,
beside the classicalHeisenberg
type
spin exchange
operators
between A.O.’s of the samesubsystem :
-
spin-exchange
operators
between A.O.’s of twoorthogonal
subsystems ;
-
angular momenta-exchange
operators ;
-
spin-
andangular momenta-exchange
operators ;
the two lasttypes
implying
necessarily
four
A.O.’sbelonging
to twoorthogonal
subsystems.
The
generalization
of theHeisenberg
effective Hamiltonianapproach
to(p/n)-filled
bands is thus inprinciple possible although
somesupplementary
dif-ficulties may arise from the existence of p electrons per
atom. This
approach
may be useful for thestudy
of thedn systems
of clusters of transition metals.Concerning
theprecise
case studiedhere,
the mainproble,m
concerned of course the restriction of ourmodel to a
single
bandthrough
the choice of a modelspace
spanned by
products
of atomicS2p
configurations.
Thehybridization
(i.e.
the involment ofsp2
atomicconfigurations)
shouldplay a
veryimportant
role,
and this is clear from the existence of a lowlying
quintet
state
503A3u-
ofB2,
which cannot enter our model. The factthat we used the exact
eigenenergies
ofB2
for thespectral
definition ofHeft
has takenimplicitly
into account most of thehybridization mixing.
Onemight
have defined the
target
space S asspanned
by
theeigenvectors
of Hhaving
thelargest
components
on themodel space
So,
some of thembeing
higher
in energy.Attempts along
this line failed togive
reliable resultsexcept
for a few almostnon-hybridized
states(the
ground
state wascorrectly predicted
to be2IIu).
Thesuccess of our model for
B3
iscertainly
linked to thefact that the
strongly
hybridized
states go tohigher
energies
on linear chains atleast,
since the lowestessentially hybrid
state6IIu
appearsonly
in9th
position
in the MO-CI calculation.
Such success would be
unlikely
for bi- andtridimen-sional clusters. We intend to
apply
our effectiveHamil-tonians to the
prediction
of themagnetic
character of linearBn
chains.It is clear that the main further
step
in the construc-tion of effective Hamiltoniansspanned by
all neutralV.B. structures would consist in
explicitly treating
thehybridization,
i.e. severalbands,
spanning
the model spaceby
all thepossible products
of neutralground
and excited valence atomicconfigurations.
Thisstrategy,
now under
study,
faces threetypes
ofproblem ;
- the
large
size of the model space makes the abinitio extraction
difficult ;
the use of therecently
proposed
intermediate Hamiltonians[23]
may solvesome intruder state
problems, necessarily occurring
insuch
situations ;
- effectiveintegrals
will be numerous, and theirhandling
inlarger
problems
may become tedious. Someanalytic fittings
of effectiveintegrals
(in
terms ofoverlap
forinstance)
may become necessary ;- the number of neutral V.B. determinants for
a
moderate-size cluster or molecule will be
large
and thediagonalization
of thegeneralized Heisenberg
Hamil-tonian matrix may become rather
![Fig. 2. - MO-CI [xx, yy, zz] -symmetry potential curves of B2. L1 states, degenerate with those of the [xy ] -symmetry](https://thumb-eu.123doks.com/thumbv2/123doknet/14220301.483578/5.892.122.456.714.1094/fig-mo-symmetry-potential-curves-states-degenerate-symmetry.webp)
![Fig. 4. - Structure of the V.B. matrix spanned by the non hybridized, Sz = 0, determinants in the [xx, yy, zz] -symmetry.](https://thumb-eu.123doks.com/thumbv2/123doknet/14220301.483578/7.892.481.830.107.277/fig-structure-matrix-spanned-non-hybridized-determinants-symmetry.webp)


![Fig. 7a. - MO-CI [xyz] - and [zzz]-symmetry lowest potential curves of B3.](https://thumb-eu.123doks.com/thumbv2/123doknet/14220301.483578/14.892.77.408.106.579/fig-mo-ci-xyz-symmetry-lowest-potential-curves.webp)