Enhancer–gene maps in the human and zebrafish genomes using evolutionary linkage conservation
Texte intégral
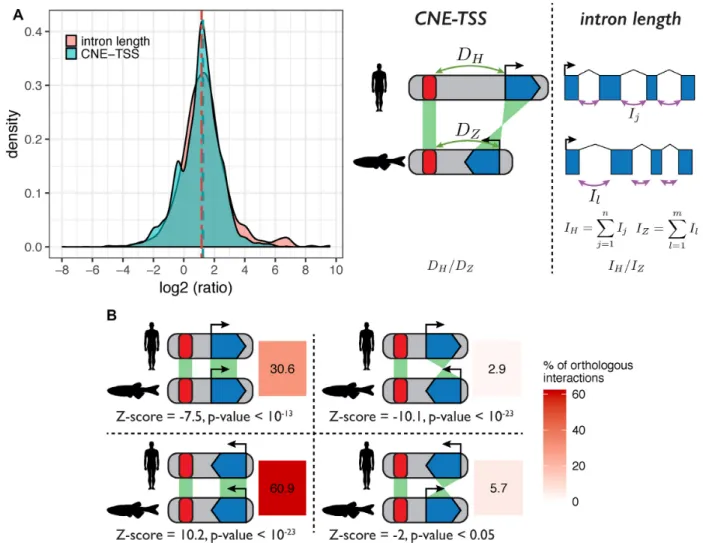
Figure

Documents relatifs
The New Zealand Immigration and Protection Tribunal made a point of saying that the effects of climate change were not the reason for granting residency to the family, but
where we made explicit the dependence on µ and on the
FIG 2 Distribution of the diversity numbers of antibiotic resistance and virulence factor protein families in human gut metagenomes versus metagenome protein family richness..
Immunohistochemistry experiments with anti-Cyp19a1 antibodies revealed that in control and E 2 -exposed fish testis (7 and 14 days), Cyp19a1a immunoreactivity was observed
xylanisolvens XB1A T grown on insoluble oat-spelt xy- lan (OSX) at mid- and late-log phase relative to glucose and/or xylose (Table 1) (Additional file 1: Figure S1)..
These crises may be the disruptive entries of newcomers into a tightly regulated market.. Most of the time, these bring with them a new technology or a radically
To test if the identified puta- tive bovine enhancer regions are enriched with functional variants that affect milk production traits, we performed genome-wide association
Then, we used whole‑ genome sequence data from three pig lines to calculate: a variant intolerance score that measures the tolerance of genes to protein coding variation; an
