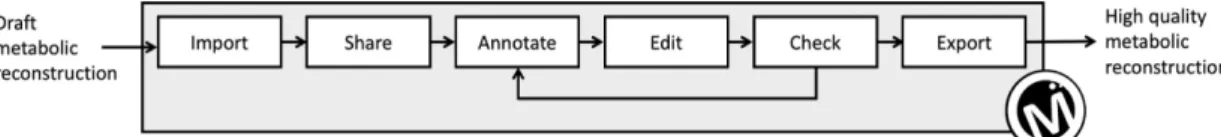
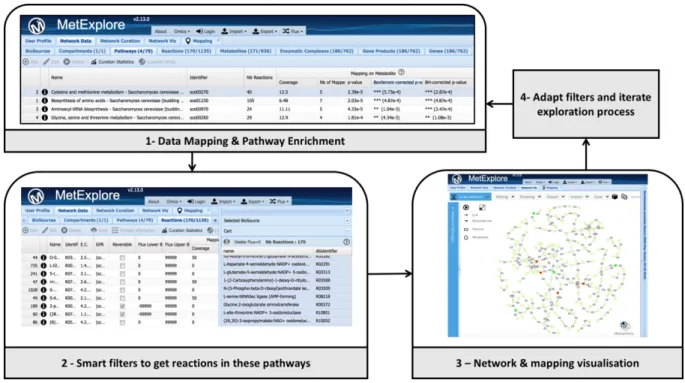
MetExplore: collaborative edition and exploration of metabolic networks
Texte intégral
Figure

Documents relatifs
TOPOLOGICAL ANALYSIS by STATISTICAL TOOLS and GRAPH THEORY !... Main
The increased arginine concentration in POAG plasma was evidenced by three of the four data methods and was already reported in the targeted metabolomics study [7], together with
Before attempting to prove the results, it is comforting to perform a few easy checks so as to ensure that these results seem correct beyond reasonable doubt (cf the end of section 1
By combining regulatory sequence analy- sis and metabolic pathway analysis, one could obtain two independent and comple- mentary sources of information for these clusters
Whenever a query is formulated selecting for instance association rules from these tables, a run of a data mining algorithm is triggered (e.g., Apriori [6]) that computes the result
Similarly, if P3 is an exogenous process (it occurred previously because the rare stimulus was more intense), then it should now also be larger to the more intense
To prepare data for use, we divide a video dataset into clips, and we compute a signature for each clip using an ensem- ble of deep learning neural networks.. Currently, we define
TE-rich regions, which encompass one third of the genome, are enriched in putative effector genes that present the same expression pattern (no or a low expression level during
