Full Terms & Conditions of access and use can be found at
https://www.tandfonline.com/action/journalInformation?journalCode=kccy20
Cell Cycle
ISSN: 1538-4101 (Print) 1551-4005 (Online) Journal homepage: https://www.tandfonline.com/loi/kccy20
Budding yeast 14-3-3 proteins contribute to
the robustness of the DNA damage and spindle
checkpoints
Nathalie Grandin & Michel Charbonneau
To cite this article: Nathalie Grandin & Michel Charbonneau (2008) Budding yeast 14-3-3 proteins contribute to the robustness of the DNA damage and spindle checkpoints, Cell Cycle, 7:17,
2749-2761, DOI: 10.4161/cc.7.17.6592
To link to this article: https://doi.org/10.4161/cc.7.17.6592
Published online: 01 Sep 2008.
Submit your article to this journal
Article views: 238
View related articles
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
Cells respond to DNA or mitotic spindle damage by activatingspecific pathways that halt the cell cycle to allow for possible repair. Here, we report that inactivation of one of the Saccharomyces
cere-visiae 14-3-3 proteins, Bmh1, as well as the bmh1-S189P bmh2
mutant, failed to exhibit normal spindle damage-induced cell cycle delay and conferred hypersensitivity to benomyl or nocodazole. These defects were additive with those conferred by the bub2 and
mad2 spindle checkpoint mutations. Following cdc13-1-induced
DNA damage, the 14-3-3 response was additive with those provided by the Mec1 (ATR-related)-controlled Rad53 (CHK2-related) and Chk1 (CHK1-(CHK2-related) checkpoint pathways and also distinct from the PKA (Protein Kinase A)-controlled response. Therefore, the budding yeast 14-3-3 proteins contribute to the robustness of the two major mitotic checkpoints and, by doing so, may also ensure optimal coordination between the responses to two distinct types of damage.
Introduction
Faithful transmission of genetic information from one generation to the next requires perfect stability of the genome. Following dupli-cation of the genetic material during DNA replidupli-cation, very accurate segregation of the chromosomes on the microtubule spindle during mitosis is required. These events are controlled by specific checkpoint mechanisms that act to signal damage, activate repair and halt the cell cycle as long as the damage persists. The two major G2/M cell cycle checkpoints are the so-called DNA damage checkpoint, the main components of which, in the budding yeast Saccharomyces cerevisiae, are Mec1, Mec3, Rad53 and Chk1, and the spindle checkpoint, which comprises the two independent Bub2 and Mad2 branches.15,30
In general, the components of these two pathways have been widely conserved throughout eukaryotic evolution and yeast is an excellent model system for studying checkpoint function.
Recent studies have highlighted the existence of numerous crosstalks between the various components of the DNA damage
checkpoint machinery that modulate the response depending on the location and intensity of the damage.32 Quite curiously, connections
between DNA damage checkpoint proteins and spindle checkpoint proteins were also found. Thus, treatment of budding yeast cells with nocodazole, a microtubule-depolymerizing chemical, unexpect-edly triggered activation of the DNA damage checkpoint kinase Rad53.2 Conversely, defective DNA replication in a cdc6 budding
yeast mutant activated the spindle assembly checkpoint.36 Similarly,
the budding yeast cdc13-1 mutation, which causes telomeric DNA damage, was sensed by the Bub2 branch of the spindle checkpoint, which normally monitors spindle orientation.43 In fact, the Bub2/
Bfa1 complex was subsequently recognized as a target of multiple checkpoint pathways.18 Thus, Bfa1 was inhibited by
Cdc5-depen-dent phosphorylation in two distinct manners, mediated or not by Mad2, depending on the type of spindle defect, but activated by Rad53-dependent phosphorylation following cdc13-1-induced DNA damage.18 Likewise, the DNA damage checkpoint kinase Chk1
regulates the stabilization of securin Pds1, an anaphase inhibitor,34,44
which is also a target of Mad2, the spindle checkpoint branch monitoring spindle assembly, kinetochore attachment and tension defects.8,30 Further adding to this complexity, the APC (Anaphase
Promoting Complex) activator Cdc20, whose primary effector is Mad2,27 was also the target of cdc13-1-induced DNA damage
sensed by PKA (Protein Kinase A).35 The connections between the
DNA damage and spindle checkpoints are not restricted to
cdc13-1-activated events. Indeed, for instance, Mad2 contributed to the
cell cycle arrest induced by mutations in various DNA replication proteins.7 In the absence of Mec1, Mad2 also partially contributed
to the G2/M arrest of UV-irradiated cells.3 Even more surprisingly,
in cells treated with DNA replication inhibitors, Mec1 and Rad53 controlled chromosome segregation, not by inhibiting entry into mitosis as expected, but by directly regulating spindle dynamics.21
This surprising and sophisticated interdependency of the DNA damage and spindle checkpoints probably arose during evolution in part because they both function to block the metaphase/anaphase transition and the exit from mitosis.
With many other examples, not only in yeast, but also in Drosophila and mammals (reviewed in refs. 5, 19, 33 and 37), it is clear that the DNA damage and spindle checkpoints are tightly connected. The nature of the defects and the way they are sensed sometimes give clues concerning these connections. For instance, a defect in chromosome organization can understandably confer a
*Correspondence to: Michel Charbonneau; Ecole Normale Supérieure de Lyon; UMR CNRS 5239; 46, allée d’Italie; Lyon 69364 France; Tel.: 33.472.72.81.70; Fax: 33.472.72.80.80; Email: Michel.Charbonneau@ens-lyon.fr
Submitted: 04/07/08; Revised: 07/14/08; Accepted: 07/14/08 Previously published online as a Cell Cycle E-publication: http://www.landesbioscience.com/journals/cc/article/6592
Report
Budding yeast 14-3-3 proteins contribute to the robustness of the DNA
damage and spindle checkpoints
Nathalie Grandin and Michel Charbonneau*
IFR128 BioSciences Gerland; UMR CNRS/ENS n° 5239; Ecole Normale Supérieure de Lyon; Lyon, France
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
defect in kinetochore attachment if concerning centromeric DNA, as observed previously.29 Very rarely only, a checkpoint protein can
act simultaneously in both types of checkpoint. This was the case for Mec1 and Rad53, as seen above. Likewise, treatment of cells with nocodazole activated both Rad53 and its effector, Rad9, in a Mad2-dependent manner but inMad2-dependently of the other components of the DNA damage checkpoint.2 Moreover, the rad53 and rad9 null
mutations were not hypersensitive to spindle dysfunction.2
Here, we report that mutations in budding yeast BMH1, one of the two 14-3-3 budding yeast genes,42 confer hypersensitivity to
both DNA damage and spindle damage and failure to delay cell cycle progression in response to both types of damage. This study began with the isolation of a mutation in BMH1 as a partial suppressor of the temperature sensitivity of the cdc13-1 mutation. The cdc13-1 mutation, which confers a defect in telomere end protection,9 has
been used to isolate mutations in the DNA damage checkpoint.45,46
In fact, recently, a bmh1 null mutation was also isolated in such a
cdc13-1 genetic screen,4 just as we report here. Bmh1 has already
been implicated in the response to DNA damage25,39 and one of the
seven 14-3-3 human isoforms, induced by DNA damage, is silenced in carcinomas.16 During the course of the present study, we made the
surprising observation that the bmh1 null mutant was hypersensitive to microtubule-depolymerizing drugs. We also report that Bmh1 is needed to impose a cell cycle delay in response to nocodazole treat-ment. The present data also show that mutations in BMH1 aggravate the response to spindle damage in cells bearing mutations in either one or both of the two major spindle checkpoint genes, BUB2 and
MAD2. In parallel, we further investigated the role of Bmh1 in the
G2/M DNA damage checkpoint and, importantly, find that it acts in a pathway that is distinct from both the Rad53, the Mec1-Chk1 and the Protein Kinase A-Cdc20 pathways.34,35,46 This dual
participation of 14-3-3 proteins in the two major mitotic check-points may indicate a safety pathway ensuring consolidation of the basal checkpoint mechanisms.
Results
Bmh1 contributes to the maintenance of cell cycle arrest after spindle damage. Bmh1 and Bmh2 are the two members of the highly
conserved 14-3-3 protein family in budding yeast. Inactivation of both genes is lethal in most strain backgrounds, including ours, while deletion of either one has no impact on cell growth, at least under standard culturing conditions. Following application of sub-lethal doses of microtubule-depolymerizing drugs, a small percentage of nocodazole-treated bmh1Δ cells initiated re-budding, a typical response in bub2Δ or mad2Δ mutants, unlike wild-type cells and cells mutated for a component of the Mec3-Ddc1-Rad17 clamp that all marked complete cell cycle arrest (Fig. 1A). The percentage of re-budding in the bmh1Δ mutant, assessed after 4 hr of treatment with 15 μg/ml of nocodazole, was 20.1% (3.3 SD, n = 8) versus 4.5% (1.5 SD, n = 6) in the wild-type. This difference was statisti-cally significant (p = 0.0008 in a paired t test).
The spindle checkpoint pathway comprises two parallel and sepa-rate branches. The Mad2 branch monitors the attachment of bipolar microtubules to the kinetochores of sister chromatids or kinetochore malfunction, as well as spindle assembly. The Bub2/Bfa1 branch monitors spindle misorientation. Although both branches are some-what interconnected,18 they nevertheless function independently
from each other.8,38 In the re-budding assay, the phenotype of
mad2Δ bmh1Δ appeared to be aggravated compared with that of mad2Δ, at all time points following treatment with nocodazole,
while, on the contrary, the percentages of re-budding were lower in the bub2Δ bmh1Δ double mutant than in the bub2Δ mutant (Fig. 1B). The differences between the mad2Δ bmh1Δ and mad2Δ strains were statistically significant, while those between the bub2Δ bmh1Δ and bub2Δ strains were significant only at the 4-hr time point (Fig. 1B).
In the continuous presence of benomyl, the viability of bmh1Δ cells was reduced compared with the wild type, becoming apparent above 14 μg/ml (Fig. 1C). Loss of viability in benomyl was slightly aggravated in bub2Δ bmh1Δ, compared with bub2Δ, as well as in
mad2Δ bmh1Δ compared with mad2Δ (Fig. 1C). Viability of the bub2Δ mad2Δ bmh1Δ triple mutant in benomyl was not aggravated
compared with that in the bub2Δ bmh1Δ and mad2Δ bmh1Δ double mutants and slightly aggravated compared with bub2Δ mad2Δ double mutant (Fig. 1C). The bub2Δ mad2Δ double mutant was surprisingly less sensitive than the mad2Δ single mutant (Fig. 1C), as observed previously.6 In contrast to wild-type cells that maintained
an arrest with a 2C DNA content, a substantial percentage of the
bub2Δ cells progressed into mitosis and accumulated with a 4C
DNA content (Fig. 1D). Although the phenotype of mad2Δ cells was not very marked under these conditions, accumulation of cells with a 4C DNA content in the bub2Δ mad2Δ double mutants was much increased compared to bub2Δ mutant alone (Fig. 1D). This increase in ploidy, attributed to additional rounds of DNA replication in the absence of cytokinesis,6 was also more severe in mad2Δ bmh1Δ
than in the corresponding single mutants, but similar in bub2Δ and
bub2Δ bmh1Δ (Fig. 1D). Noticeably, the highest percentage of cells
with a 4C DNA content was in the bub2Δ mad2Δ bmh1Δ triple mutant (Fig. 1D).
In summary, genetic inactivation of BMH1 confers benomyl/ nocodazole hypersensitivity, partially overrides the cell cycle arrest provoked by these drugs and mimics the genome instability defect conferred by the bub2Δ mutation when Mad2 is absent. However, clearly, Bmh1 does not have the same targets as Bud2 or Mad2 to arrest the cell cycle upon damage.
The S189P and S180P Bmh1 mutations confer defects in both the DNA damage and spindle checkpoints. Bmh1 plays a more
important role than Bmh2 in all the processes in which they have been implicated, as deduced from the more severe phenotypes of deleting
BMH1 than BMH2. This probably explains that a deletion of BMH1
conferred a deficiency in the mitotic checkpoints even in the presence of BMH2, as observed here. To allow further biochemical analysis of defects conferred by loss of BMH function originating from a single gene product, we set out to isolate bmh1 checkpoint mutants in the
bmh2 null background. Indeed, because the functions of Bmh1 and
Bmh2 are redundant, the bmh1Δ bmh2Δ double mutation being lethal, it was important to analyze loss of functions of Bmh1 in the absence of Bmh2. Benomyl-sensitive bmh1 mutants were sought. Twenty such mutants were isolated among ~8,000 PCR-mutagenized cells. Among the four that remained after re-checking the benomyl-sensitivity phenotype, bmh1-23 was selected for further analysis because it had the most severe phenotype. bmh1-23 sustained viability at a single, chromosomal, copy in the bmh2Δ background and was found upon sequencing to introduce a single mutation, S189P.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
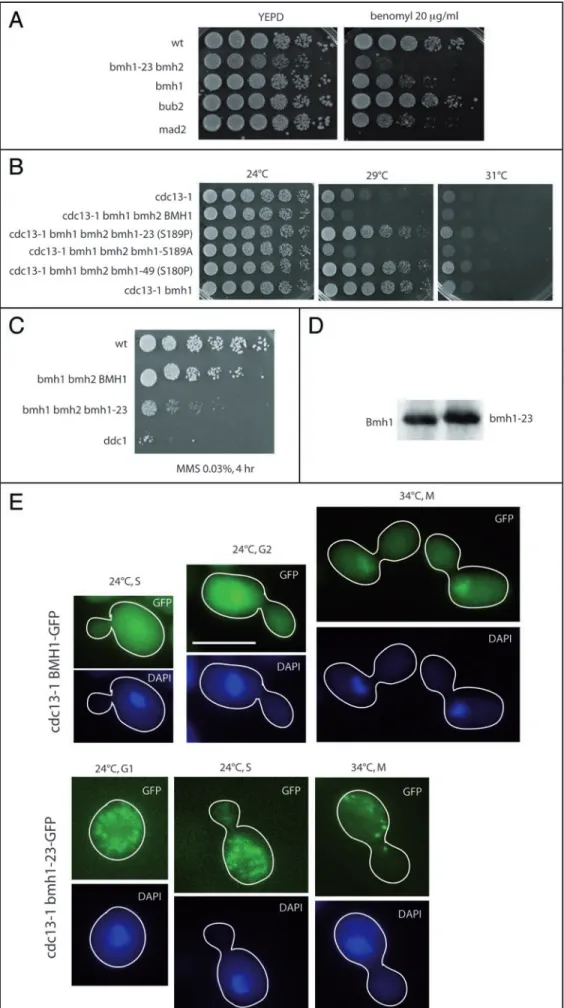
bmh1-23 mutant cells were not only hypersensitive to benomyl
(Fig. 2A), but also exhibited a defect in imposing complete mitotic cell cycle delay in response to nocodazole (data not shown). bmh1-23 also partially rescued the cdc13-1 mutation, just like the bmh1Δ mutation (Fig. 2B), as observed before4 and bmh1-23 mutant cells
were also hypersensitive to MMS (Fig. 2C). Bmh1 serine 189 is the homologue of serine 185, phosphorylated in two human 14-3-3 isoforms.1 Neither bmh1 S189A nor bmh1 S189D, mimicking
non-phosphorylatable and constitutively phosphorylated forms, respectively, were defective in the DNA damage and spindle check-points (Fig. 2B and data not shown). Both alleles sustained viability, at single genomic copy, in the bmh2 null background. Slow growth was observed for both the bmh1 S189A and bmh1 S189D mutant strains, more pronounced for the latter, with no other apparent defect (data not shown).
Endogenous myc-Bmh1-23 exhibited slower gel electrophoretic mobility than wild-type myc-Bmh1 (Fig. 2D). This was consistent with the proline residue at position 189 potentially conferring a dramatic structural modification in the mutated protein. No further apparent change in abundance or electrophoretic mobility of myc-Bmh1 or myc-myc-Bmh1-23 was seen in cdc13-1-damaged cells (data not shown). Endogenous Bmh1-23-GFP mislocalized, either in the
cdc13-1 background at the permissive temperature for growth of
24°C (Fig. 2E) or in the CDC13+ background (data not shown). The
intracellular distribution of Bmh1-GFP and Bmh1-23-GFP did not change in cdc13-1-damaged cells, incubated for 2–4 hr at the restric-tive temperature for growth of 34°C (Fig. 2E).
Subsequently, we isolated a second bmh1 mutant, bmh1-49, also in the bmh2Δ background, that behaved like bmh1-23 bmh2Δ in the cdc13-1 background (Fig. 2C). bmh1-49 contained a single point mutation changing serine 180 to proline 180. Noticeably, this bmh1-49 mutation introduced a serine to proline change, just like the S189P mutation in bmh1-23. Since both residues are likely to be very near each other in the protein, this might suggest that a particular conformation of that region of the protein plays an essen-tial role in the 14-3-3’s checkpoint function.
Previously, it was shown that a bmh1Δ bmh2 S189P mutant conferred temperature sensitivity (at 37°C) that could be suppressed by disrupting RTG3, coding for a general transcription regulator.41
We found that the cdc13-1 bmh1Δ rtg3Δ triple mutant was still checkpoint-deficient, to the same extent as the RTG3+ sister strain
(data not shown). Therefore, the checkpoint defect conferred by
bmh1Δ, and probably, by extension, by bmh1-23, is not related to
Rtg3 functions.
cdc13-1 bmh1 K51R cells (deficient in Bmh1-ligand
interac-tion31) behaved like the cdc13-1 bmh1Δ and the cdc13-1 bmh1-23
strains at 29°C (data not shown). Therefore, formation of one of the two phosphopeptide-binding pockets of Bmh1 is essential for its ability to affect the DNA damage-induced checkpoint arrest.
Bmh1 contributes to the robustness of the DNA damage check-point. Temperature-sensitive cdc13-1 mutant cells of Saccharomyces
cerevisiae accumulate abnormal levels of single-stranded DNA at
the telomeres.9 This is perceived as a DNA damage and triggers a
cell cycle checkpoint-dependent arrest.9,45 The cdc13-1 mutation
has been used as a tool to isolate or characterize novel checkpoint proteins, including Mec1, Rad53, Mec3, Chk1 and the PKA (Protein Kinase A) complex.34,35,46 A null mutation in BMH1 was
also recently isolated in a cdc13-1-based genome-wide genetic screen and, like mutations in MEC1, RAD53 and MEC3, conferred over-riding of the damage-induced cell cycle arrest.4
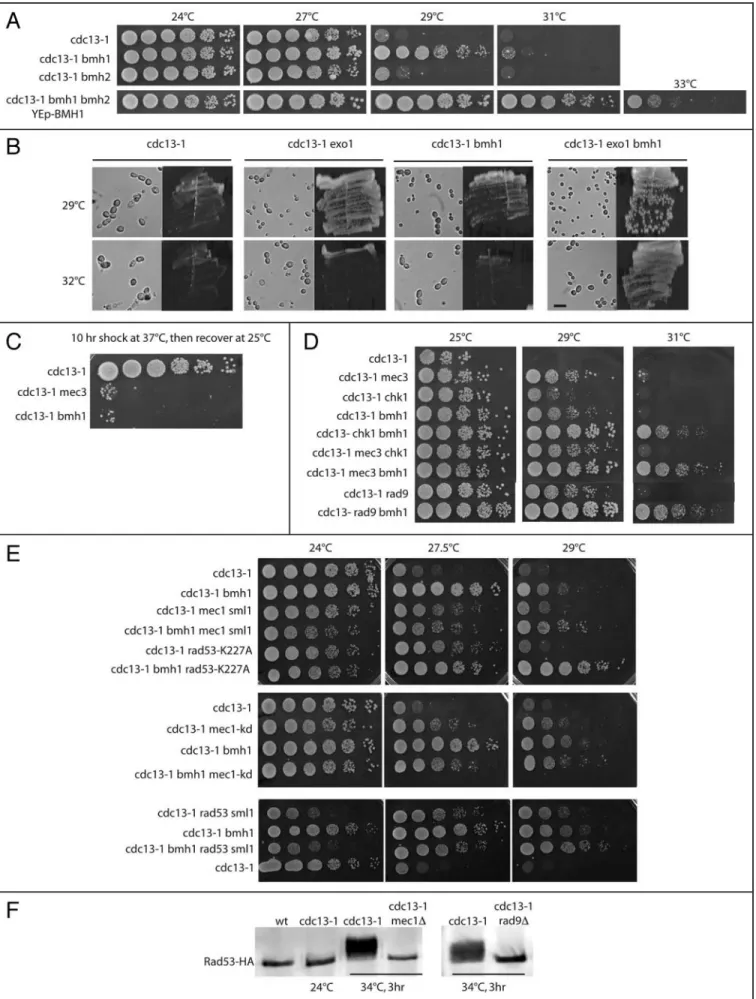
A bmh1 mutation was responsible for the growth of spontaneous revertants of cdc13-1 cells left growing at 29°C and, in a distinct screen, for that of UV-mutagenized cdc13-1 cells, also at 29°C (data not shown). At the semi-permissive temperature of 29°C, cdc13-1
bmh1Δ cells were delayed in G2/M, like cdc13-1 cells, but progressed more rapidly to the next G1, as shown by FACS analysis and accumu-lation of large-budded cells (Fig. 3A and B), suggesting that G2/M checkpoint pathways were still operational in the absence of Bmh1. At 31°C, temperature at which both cdc13-1 and cdc13-1 bmh1Δ cells arrested, the absence of Bmh1 led to a partial release of the
cdc13-1-imposed block to migration of the spindle and nucleus into
the daughter cell (Fig. 3D). However, movement of the nucleus to the neck region also occurred in cdc13-1 cells after prolonged incuba-tion at 37°C (data not shown).
Synchronized bmh1 null cells exhibited no major defect in cell cycle profile at either 24 (Fig. 3C) or 29°C (Fig. 3A), thus elimi-nating cell cycle lengthening as a possible cause for rescue. Deletion of BMH2 had only a slight effect on the growth of cdc13-1 mutants (Fig. 4A). Interestingly, cdc13-1 bmh1Δ bmh2Δ overexpressing
BMH1 (from an episomal, 2 μ, plasmid under the control of its
native promoter) were even more deficient in the cdc13-1-induced G2/M checkpoint than cdc13-1 bmh1Δ cells (Fig. 4A). Since the effect of bmh2Δ on cdc13-1 is less than that of bmh1Δ (Fig. 4A), this data suggested that overexpression of BMH1 conferred a true checkpoint defect, more severe than that conferred by disrupting either BMH1 or BMH2. Indeed, if overexpressed BMH1 was simply complementing bmh1Δ, then cdc13-1 bmh1Δ bmh2Δ YEp-BMH1 would grow up to only 29°C, like cdc13-1 bmh2Δ, which was not the case (Fig. 4A). Overexpression of BMH1 also rescued cdc13-1 in a bmh1Δ BMH2+ or BMH1+ BMH2+ background (data not shown).
These strains grew also at 33°C or below, meaning the additional
BMH proteins provided by the chromosomal copy of BMH1 or
Figure 1. bmh1 null cells fail to mark complete cell cycle arrest in response to microtubule-depolymerizing drugs. (A) Representative cell morphologies of the indicated strains show that a substantial number of bmh1Δ cells initiated re-budding, at 29°C, in medium containing 15 μg/ml nocodazole, although to a lesser extent than the bub2Δ and mad2Δ mutant. rad17Δ cells, defective in the DNA damage checkpoint, exhibited total arrest, just like the wild-type control (wt). Scale bar, 10 μm. (B) Budding index after treatment of the liquid cultures with 15 μg/ml nocodazole at 29°C. Error bars represent standard devia-tions from at least three experiments. A paired t test was used to determine significance (values were significantly different when p < 0.05). Re-budding in
mad2Δ bmh1Δ was very significantly different from that in mad2Δ at all time points (p = 0.002 after 3 hr of nocodazole treatment; p = 0.001 at 4 hr; p =
0.004 at 5 hr). In contrast, re-budding in bub2Δ bmh1Δ was significantly different from that in bub2Δ only after 4 hr of nocodazole treatment (p = 0.0025 versus p = 0.07 at 3 hr and p = 0.3 at 5 hr). On the other hand, the differences in re-budding between bub2Δ mad2Δ and bub2Δ mad2Δ bmh1Δ were not significantly different. (C) Strains of the indicated relevant genotype were grown on YEPD plates containing the indicated amount of benomyl. bmh1Δ cells were hypersensitive to benomyl, starting at 16–18 μg/ml and this sensitivity was aggravated in the absence of Bub2 or Mad2, best seen at 18 μg/ ml benomyl. Ten-fold serial dilutions (from left to right in each row) of strains were grown for 2–3 days at the indicated temperatures and photographed. (D) FACS analysis of strains with the indicated relevant genotype following treatment of the liquid cultures with 15 μg/ml nocodazole.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
Figure 2. The bmh1 S189P mutant. (A) The bmh1-23 (S189P) bmh2Δ mutant is hypersensitive to benomyl. bmh1-23 (ORF plus flanking sequences) was integrated at the TRP1 locus in a bmh1::KanMX4
bmh2::KanMX4 mutant. Ten-fold serial
dilu-tions (from left to right in each row) of strains were grown for 3 days at 24°C and photographed. (B) The bmh1 S189P and S180P bmh2Δ mutants (but not S189A) are deficient in the DNA damage checkpoint, induced by cdc13-1 at 29°C, to the same extent as the bmh1 null mutant. (C) The
bmh1-23 (S189P) mutant is
hypersensi-tive to MMS (treatment with 0.03%, 4 hr, before spotting on YEPD medium without MMS). ddc1Δ, deficient in the DNA dam-age checkpoint, was used as a control. Both the bmh1-23 and BMH1 alleles were inte-grated at the TRP1 locus in a bmh1Δ bmh2Δ strain. (D) Myc-Bmh1-23 exhibits slightly retarded electrophoretic mobility compared with wild-type myc-Bmh1. (E) Localization of Bmh1-GFP (top two rows) or Bmh1-23-GFP (bottom two rows) in living cells (both alleles were integrated at the LEU2 locus in a
cdc13-1 bmh1Δ strain) at permissive (24°C)
or restrictive (34°C) temperature for growth for cdc13-1. Scale bar, 5 μm.
BMH2, or both, did not significantly
change the checkpoint defect caused by overexpressed BMH1.
The amount of single-stranded DNA that is abnormally generated in cdc13-1 cells is taken into account during acti-vation of the DNA damage response. The EXO1 exonuclease is partially responsible for the generation of single-stranded DNA in cdc13-1 mutants.28
Thus, in cdc13-1 cells bearing a null mutation in EXO1, there is less single-stranded DNA generated, hence less damage perceived and, consequently, these cdc13-1 exo1Δ mutant cells grow better than cdc13-1 EXO1+ cells (Fig.
4B).28 Here, we show that the
dele-tion of BMH1 does not rescue cdc13-1 by effecting Exo1 (Fig. 4B). Indeed,
cdc13-1 exo1Δ bmh1Δ cells grew better
than cdc13-1 exo1Δ or cdc13-1 bmh1Δ cells (Fig. 4B), indicating that Exo1 and Bmh1 act in two distinct path-ways to rescue cdc13-1 temperature sensitivity.
cdc13-1 bmh1Δ cells recovered
poorly from a 10 hr-shock at 37°C, a phenotype similar to that in the DNA damage checkpoint-deficient cdc13-1
mec3Δ mutant (Fig. 4C). This result
strongly suggests that the diminution of cdc13-1 temperature sensitivity
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
Figure 3. For figure legend, see page 7.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
observed after deletion of BMH1 operates in the same way as mutations in checkpoint proteins do and further confirms that inactivation of Bmh1 does not act by diminishing the amount of
cdc13-1-induced damage.
Mec3, a DNA damage sensor, is required for activation of Mec1, another sensor, as well as for that of Rad53 and Chk1, two effector kinases in complex with the Rad9 adaptor acting in parallel path-ways.15,32 Inactivating BMH1 in cdc13-1 mec3Δ, cdc13-1 chk1Δ or
cdc13-1 rad9Δ mutants resulted in an additive effect, best visible at
31°C (Fig. 4D). In contrast, cdc13-1 mec3Δ chk1Δ triple mutants did not grow better at 29 or 31°C than cdc13-1 mec3Δ mutants (Fig. 4D), as expected from the fact that Mec3 directly controls both the Chk1 and Rad53 pathways.32 Similar additive effects were
visible at 29°C after combining the bmh1 null mutation with rad53 or mec1 mutations in the cdc13-1 background (Fig. 4E). However, interpretation of the results from the experiments using mec1 and
rad53 mutations was obscured by the existence of a synthetic growth
defect between cdc13-1 and these mutations. Indeed, at 24°C, all the strains harvesting cdc13-1 and either a rad53 or a mec1 mutation were impaired in their growth capacities (Fig. 4E, left). For instance the cdc13-1 rad53Δ sml1Δ strain grew much worse than the cdc13-1 strain at 24°C (the synthetic effect being also detectable at 27.5°C), but yet experienced improved growth over the latter at 29°C (Fig. 4E, bottom). In fact, clearly, all the cdc13-1 rad53 mutants (rad53Δ
sml1Δ or rad53-K227A) and cdc13-1 mec1 mutants (mec1Δ sml1Δ or mec1-kd) exhibited improved growth at 29°C, the lowest restrictive
temperature at which the loss of checkpoint function is currently assessed in cdc13-1 mutants,9 after BMH1 had been inactivated (Fig.
4E). Both MEC1 and RAD53 are essential genes and their inactiva-tion, even in the absence of SML1 (that alleviates the lethality caused by their inactivation), is known to greatly affect DNA replication. Since Cdc13 has been implicated in telomeric DNA replication,13
synthetic growth defects are likely to be expected. The larger addi-tional checkpoint effect seen with rad53-K227A (compared with
rad53Δ sml1Δ) is very likely explained by the fact that this mutation
is not lethal, Rad53-K227A still assuming its essential DNA replica-tion funcreplica-tion. Most importantly, given problems in interpreting the data with the rad53 and mec1 mutations due to synthetic growth defects with cdc13-1 as explained above, it should be noted that Rad9 is essential for operation of both the Rad53-Rad9 and Chk1-Rad9 pathways15,32 and that the other branch of the Rad53 pathway,
Rad53-Mrc1, is not activated upon cdc13-1-induced damage.13
Consistent with this, Rad53 activation was totally lost after RAD9 had been genetically inactivated, similarly to what happens when Mec1, functioning upstream of Rad53, is absent (Fig. 4F). Moreover, Mec3 is essential for operation of the Mec1-Rad53 pathway15,32 and,
as expected, inactivation of MEC3 also resulted in complete loss of Rad53 activation (data not shown). In summary, analysis of these
cdc13-1 mutants strongly suggests that Bmh1 functions in a DNA
damage checkpoint that is distinct from the Mec1-Rad53-Rad9 and Mec1-Chk1-Rad9 pathways.
It has been well established that both the RAD53 and CHK1 checkpoint kinases exhibit a DNA damage-dependent electrophoretic shift. These upper shifted bands of Rad53 and Chk1 are the activated forms of the proteins corresponding to activation of kinase activity by phoshorylation (see, for instance, ref. 25 and 34). Deletion of
BMH1 in cells experiencing cdc13-1-induced DNA damage did not
greatly affect Rad53 electrophoretic shift up (the activated form), either at semi-permissive (29°C) or restrictive temperature (34°C) for growth (Fig. 5A). We noted that in the 29°C block and release experiment (Fig. 5A, bottom), at the 120 and 135 min time points, the less phoshorylated forms of Rad53 appeared to be more intense in cdc13-1 bmh1Δ than in cdc13-1 BMH1+, suggesting a premature
inactivation of Rad53 in the absence of Bmh1. In fact, at 29°C, the damage in these cells is much less important than at 34°C, as judged by Rad53 activation (Fig. 5A, compare top and bottom), thus rendering accurate comparisons between the two strains rather uncertain. Moreover, we also note that at 29°C, cdc13-1 bmh1Δ cells re-enter a new cell cycle, while cdc13-1 BMH1+ remain blocked
at G2/M, as shown in Figure 3B above, thereby possibly altering Rad53 activation status. To circumvent this problem, we performed additional experiments in which alpha factor was added a second time after the release from the initial alpha factor block to prevent re-entry into the next cell cycle. Under these conditions, we observed that the Rad53 activation forms were very similar in the absence or presence of Bmh1 after prolonged incubation at 29°C (Fig. 5B, left). Additional experiments to address this point, consisting in analyzing Rad53 activation after prolonged incubation of cdc13-1 mutants at 31°C to elicit complete cell cycle arrest, confirmed that the pattern of activation was very similar whether Bmh1 was present or absent (Fig. 5B, right). From these experiments, we conclude that, following
cdc13-1-induced damage, the pattern of Rad53 activation is not
affected by the absence of Bmh1. bmh1 null mutants treated with the DNA damaging agent methyl methane sulfonate or with the DNA replication inhibitor hydroxyurea exhibited activated forms of Rad53 (Fig. 5C), in agreement with previous data obtained following UV irradiation.25 We noted, however, that migration of the Rad53
activated forms was different in bmh1Δ and the wild-type control (Fig. 5C). Prolonged incubation of bmh1Δ cells in MMS or HU, here 4 hr, might perturb Rad53 stability or activation, as these cells have been shown to be hypersensitive to HU.25 To avoid this possible
complication, we performed additional experiments in which cells
Figure 3. Cell cycle defects of bmh1Δ cells after cdc13-1-induced DNA damage. (A) FACS analysis of bmh1Δ and cdc13-1 bmh1Δ cells, and their BMH1+
sister cells, synchronously released into the cell cycle from an alpha-factor block in G1. The rho0 mutation was introduced to eliminate possible signals from
the mitochondrial DNA. Indeed, in the cdc13-1 mutants at 29°C, cells dramatically slow down cell cycle progression at G2/M but nevertheless continue to grow in size and, therefore, continue to accumulate mitochondrial DNA, which may affect the signals from the nucleus DNA content during FACS analysis. (B) Morphologies of alpha-factor-synchronized living cells at 29.5°C. Initiation of budding took place simultaneously in the wild-type, cdc13-1 and cdc13-1
bmh1Δ strains (arrowheads at 45 min). However, the next time frames (re-budding marked by arrowheads) illustrate the checkpoint defect in the cdc13-1 bmh1Δ cells. (C) Cell cycle distribution by FACS in isogenic wild-type and bmh1Δ cells at 24°C after synchronous release from alpha-factor block. (D) Tubulin
labeling and DAPI staining in cdc13-1 (top two rows) or cdc13-1 bmh1Δ (bottom two rows) cells. The percentages of cells with the spindle and nucleus in the mother cell or in both the mother and daughter cells, as schematically indicated, at the 31°C arrest were calculated after observation of 425 cdc13-1 and 562 cdc13-1 bmh1Δ dividing cells. The criteria used to distinguish between DNA present only in the mother cell versus DNA present in both the mother and daughter cells are as follows. A round DAPI-stained nucleus is indicative of DNA present only in the mother cell. In contrast, as soon as the nucleus starts migrating in the daughter cell, it loses its round shape because it is constrained by the bud neck. Scale bar, 5 μm.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
were synchronized with alpha factor and MMS-induced Rad53 activation analyzed during the course of a single cell cycle. Under these conditions, Rad53 was activated with similar kinetics in both samples (Fig. 5D). Because activation of Chk1 was affected in some UV-irradiated bmh1 bmh2Δ mutants,25 we analyzed its activation
during cdc13-1-induced damage in the presence or absence of Bmh1. Activation of Chk1 took place in synchronized cdc13-1 bmh1Δ cells, at 29 or 34°C, to the same extent as in control cdc13-1 BMH1+
cells (data not shown). This result confirms the previously observed absence of an effect of UV irradiation on Chk1 activation in bmh1Δ
BMH2+ cells.25
Bub2, but not Mad2, is activated upon cdc13-1-induced damage.43 Inactivating either BUB2 or BMH1 in cdc13-1 mutants
led to distinct phenotypes. Noticeably, cdc13-1 bub2Δ cells formed microcolonies at 29°C, while cdc13-1 bmh1Δ cells continued to slowly divide without forming microcolonies (Suppl. material, Fig. S1). Tpk1, a component of the PKA complex participates in
cdc13-1-induced checkpoint response.35 A genetic analysis showed
that bmh1Δ and tpk1Δ act in two distinct ways in cdc13-1 rescue (Suppl. material). The main argument was that, at 29°C, cdc13-1 mutants in which both TPK1 and CHK1 had been inactivated (the condition required for observation of the PKA checkpoint function)35 exhibited improved growth after BMH1 had been
inac-tivated (Suppl. material, Fig. S2A). Further, the Bmh1-1 protein, defective in binding Ste20, a mitotic inducer implicated in the exit of mitosis, was still proficient in checkpoint function (Suppl. mate-rial, Fig. S2B) and, in addition, cdc13-1 bmh1Δ grew like cdc13-1
bmh1Δ ste20Δ (Fig. S2C). Cdc20, an activator of APC (Anaphase
Promoting Complex), represents the major target of PKA check-point function.35 We find here that the stability of Cdc20, as well as
that of its target, Pds1 securin (the destruction of which is required for chromosome segregation), were not modified in the absence of Bmh1 (Suppl. material, Fig. S3A and B), thereby confirming the data above. Finally, cdc13-1 rescue by bmh1Δ did not appear to act through Acm1, recently identified in a complex with Cdh1 and Bmh1 (Suppl. material). Indeed, cdc13-1 acm1Δ grew exactly like
cdc13-1 ACM1+ cells, unlike cdc13-1 bmh1Δ cells (data not shown),
a result not expected if Acm1 was acting together with Bmh1 to maintain Cdh1, another APC activator, inactive upon DNA damage (Suppl. material).
Inactivation of Bmh1 does not affect Clb2 levels. cdc13-1 bmh1Δ
and cdc13-1 BMH1+ cells progressing synchronously at the
restric-tive temperature of 34°C following release from alpha-factor block
exhibited identical kinetics of accumulation of Clb2, the major mitotic cyclin in budding yeast, and no premature degradation (Fig. 6).
Discussion
The present data lead us to propose that a previously unsuspected role of the budding yeast 14-3-3 proteins is to contribute to the robustness of the DNA damage and spindle checkpoints. 14-3-3 proteins are very abundant proteins12 that regulate a remarkable
amount of protein-protein interactions in various cellular functions, such as intracellular trafficking/targeting, signal transduction, the regulation of transcription, control of cytoskeletal organization, apoptosis, regulation of the cell cycle, including the control of DNA damage checkpoints. 14-3-3 proteins are able to bind numerous proteins, mostly phosphorylated proteins, resulting in inhibition or activation of the partner protein. 14-3-3 binding may result in a change in the subcellular localization, a change in the interac-tion of the binding partners with other proteins or to shielding of binding sites.1,26,40,42 In budding yeast, a single recent proteomic
study, based on multistep immunoaffinity purification and mass spectrometry, identified 271 proteins binding Bmh1/2 in a phos-phorylation-dependent manner.20 Other genome-wide analyses,
also in budding yeast, identified around sixty proteins physically interacting with Bmh1/2, using similar techniques.10,11,17,22 Many
other physical interactions with Bmh1/2 have also been identified punctually and a vast number of mutations exhibiting aggravated phenotypes in combination with the bmh null mutations have been found (see references at Saccharomyces Genome Database website, http://db.yeastgenome.org). It is therefore probable that the Bmh1/2 proteins, which function as molecule adaptors, control an intricate network of mitotic proteins through physical contacts modulated in a highly complex manner by the various reactions of phosphorylation and dephosphorylation that take place during mitosis.
Intriguingly, given the vast amount of data accumulated on 14-3-3 proteins from various organisms, to our knowledge, evidence for 14-3-3 mutant proteins conferring hypersensitivity to microtubule-depolymerizing drugs or a defect in cell cycle delay in response to spindle damage had never been reported. Thus, although it is clear that 14-3-3 proteins function in the DNA damage checkpoint and have been directly linked to cancer and apoptosis,16,47 evidence for
inter-actions with the spindle checkpoints was still lacking. Importantly, however, we are not saying that the budding yeast 14-3-3 proteins are part of the spindle checkpoint but, rather, that they functionally interact with the two main branches of that checkpoint, the Bub2 and
Figure 4. Bmh1 and induced telomeric DNA damage. (A) Unlike that of BMH1, inactivation of BMH2 had basically no effect on the cdc13-1-induced DNA damage checkpoint, while overexpression of BMH1 (from an episomal plasmid, under the control of native promoter) had a much stronger effect than the bmh1Δ mutation. (B) Bmh1 does not act in the Exo1 pathway. Deletion of EXO1 results in the decrease of cdc13-1-induced DNA damage.28
At 29°C, cdc13-1 cells are bigger (and grow slower) than cdc13-1 exo1Δ cells because damage is more important in the former (cells continue to grow in size when slowed down or arrested, here by the checkpoint, at G2/M).45 Deletion of BMH1 has a similar apparent effect. However, even stronger relief of
DNA damage-induced slow growth in cdc13-1 exo1Δ bmh1Δ cells (smaller cells, faster growth) compared with the other two double mutants demonstrates that Bmh1 does not act in the Exo1 pathway. Cells taken from exponentially growing cultures were photographed. Bar represents 10 μm. (C) Temperature-sensitive otherwise wild-type cdc13-1 cells efficiently recovered from a transient 37°C shock, but not when either BMH1 or MEC3 have been deleted. (D) Growth characteristics of various mutants combining the temperature-sensitive cdc13-1 mutation and non temperature-sensitive, null mutations in MEC3,
BMH1, CHK1 or RAD9. Ten-fold serial dilutions (from left to right in each row) of mutant strains were grown for 2–3 days at the indicated temperatures
and photographed. (E) Using the cdc13-1 assay illustrated above, mutations in either RAD53 (rad53-K227A, a kinase deficient allele, or a null mutation combined with the sml1Δ mutation to suppress the lethality conferred by rad53Δ) or MEC1 (mec1-kd, a kinase deficient allele, or mec1Δ sml1Δ) and bmh1Δ had additive effects at 29°C. (F) Upon damage induced by cdc13-1 at 34°C, Rad53 undergoes extensive phosphorylation, under the form of several bands migrating slower than the unactivated, non phosphorylated band. Mec1 and Rad9 are both required for Rad53 activation. At the permissive temperature of 24°C, cdc13-1 does not activate Rad53, like in the wild type (wt).
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
Figure 5. Activation of the RAD53 protein kinase is not affected in cells deleted for BMH1. (A) Activation of Rad53-HA was followed in parallel in cdc13-1 and cdc13-1 bmh1Δ cells (left and right lane, respectively, at each time point) synchronized in G1 by alpha-factor and released at 34°C (top) or 29°C (bot-tom) to induce DNA damage (provoked by the temperature-sensitive cdc13-1 mutation). Although Rad53-shifted bands appeared slightly earlier in cdc13-1
bmh1Δ than in cdc13-1 cells, at 60 min after release from the G1 block, consistent with the slightly accelerated transition from the G1 to the S phase seen by FACS analysis in the former cells (see Fig. 3A and C), these phosphorylated bands, as well as additional higher shifted bands, were similar in intensity in both samples. Note that at 29°C, in both samples, the extent of Rad53 phosphorylation (corresponding to the slower migrating forms) was smaller than at 34°C, due to the smaller damage at 29°C. (B) The level of Rad53 activation (3 hr after release at the semi-permissive temperature for cdc13-1 of 29°C) was similar in cdc13-1 and cdc13-1 bmh1Δ mutants prevented from entering a second cell cycle by re-addition of alpha factor (left), as well as following complete arrest (3 hr at 31°C, right). (C) Activation of Rad53-HA in wild-type (wt) and bmh1Δ asynchronous cells (left and right lane, respectively, in all three conditions) following a 4-hr treatment with MMS or HU, or no treatment (left). (D) Kinetics of activation of Rad53 in alpha factor-synchronized cdc13-1 and cdc13-1 bmh1Δ mutants (at the permissive temperature of 24°C) treated with 0.1% MMS.
Mad2 pathways. Indeed, since the molecular mechanism at the origin of the present observations has not been elucidated yet, one cannot conclude that Bmh1/2 define a new branch of the spindle checkpoint. In addition, we do not know yet what defects exactly Bmh1/2 can recognize in the cell, besides the damage caused by benomyl or
nocodazole. Until now, the relatively minor participation of Bmh1 in the cell cycle arrest provoked by spindle damage has prevented us from establishing genetic interactions between bmh1Δ and mutations causing specific dysfunctions, such as in spindle assembly or misori-entation, or kinetochore attachment, for instance.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
The most striking spindle checkpoint phenotype of bmh1Δ cells was that of over-replication seen in combination with
MAD2 disruption. The re-replication phenotype of the bub2Δ
and mad2Δ mutants might result from the untimely destruc-tion of Clb2 which, in turn, would lead to the activadestruc-tion of Clb5/6-Cdc28 and progression through S phase. The aggravated phenotype of the bub2Δ mad2Δ double mutant probably reflects the fact that both APCCdc20 and APCCdh1 contribute to Clb2
destruction, sequentially, in an additive manner. Bmh1 may act in a different manner to consolidate the spindle checkpoint, as endogenous levels of the major mitotic cyclin, Clb2, did not appear to be affected by deletion of BMH1. Moreover, it is not clear at the moment whether the control by Bmh1/2 of several key genes required for the G1/S transition, and hence for DNA replication,24 has something to do with the over-replication seen
here in mad2Δ bmh1Δ mutants.
Although budding yeast 14-3-3 proteins have been recently implicated in the DNA damage checkpoint,25,39 the present data
brings a major additional contribution to the matter. Indeed, we demonstrate here that Bmh1 affects the response to the cdc13-1 damage in the absence of major components of the Mec1 machinery or of more minor pathways. Based on classical genetic arguments, as well as on several studies having used the cdc13-1 mutation as a basis for genetic analysis (reviewed in refs. 4, 9, 28, 34, 35, 43, 45 and 46), the Bmh1-controlled checkpoint response functioned in a pathway distinct from those controlled by the DNA damage sensors Mec1 and Mec3. Paradoxically, while inactivation of a DNA damage checkpoint gene renders wild-type cells hypersensitive to genotoxic agents, it leads to improved growth in cdc13-1 cells.9 In fact, at the
restrictive temperature of 36°C, cdc13-1 mutant strains bearing a mutation in a DNA damage checkpoint gene formed microcolonies of cells that soon died, but had grown enough to be visually identified
Figure 6. The absence of Bmh1 does not affect endogenous levels of Clb2-2 HA (top). Total extracts from cdc13-1 and cdc13-1 bmh1Δ cells, previously synchronized in G1 with alpha-factor and released at 34°C to induce DNA damage with the temperature-sensitive cdc13-1 mutation, were immunoprecipi-tated and immunoblotted using anti-HA antibodies. Anti-actin antibody was used for controlling sample loading. Bottom shows a FACS analysis of cell cycle distribution in the same cultures.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
and subsequently allow isolation of the mutation in the checkpoint gene.46 Although viability is globally decreased in cdc13-1 rad9Δ cells
at 28–29°C, these cells grow quite well but accumulate, with each cell division, damage at their telomeres that eventually provokes death by senescence.9,13,14
Quite surprisingly, we find that following cdc13-1-induced damage, Bmh1 functioned separately from the Mec1-Rad53-Mec3 pathway and the parallel Mec1-Rad53-Chk1 pathway.34 This was
surprising because phosphorylation of Rad53 and Chk1 were defective in bmh1 bmh2Δ mutants hypersensitive to MMS or UV irradiation and defective in the G1/S and G2/M DNA damage checkpoints.25 However, importantly, in that same study, bmh1Δ
mutants exhibited normal levels of activated Rad53 and Chk1,25
as we also find here. The cdc13-1-induced damage, sensed when
BMH1 has been deleted but BMH2 intact, might be more sensitive
to the levels of BMH protein than general genotoxic agents. Indeed, other cell cycle events controlled by Bmh1/2, such as the control of DNA replication, the G1/S transition, the actin skeleton and cell wall integrity, exhibited defects only when the more abundant of the two proteins, Bmh1, was mutated and Bmh2 lacking.23-25 In this respect,
it is interesting to note that overexpression of BMH1 also conferred checkpoint defects, as already observed following UV irradiation.25
As previously noted concerning the checkpoint defect caused by overexpression of BMH1 under the control of the inducible GAL1 promoter,25 increasing the levels of Bmh1 might seriously unbalance
the association of Bmh1 with one or several of its numerous targets, thereby possibly affecting the other DNA damage checkpoints. In another study, Bmh1/2 physically interacted with all phosphorylated forms of Rad53 produced upon MMS-induced DNA damage,39 yet
appear to function in a separate pathway when damage is induced by the cdc13-1 mutation, as shown here. We propose that the physical interaction taking place between Rad53 and Bmh1/2 following MMS treatment39 represents a connection between two parallel
pathways of detection of DNA damage but that, nevertheless, Rad53 and Bmh1/2 clearly have distinct targets.
The cdc13-1-induced damage also activates the Bub2 branch of the spindle chekpoint,28,43 events that correlated with Rad53-dependent
phosphorylation of Bfa1.18 However, clearly, the phenotype of
cdc13-1 bub2Δ or cdc13-1 bfa1Δ was different from that of cdc13-1 bmh1Δ. By affecting telomeres, the cdc13-1 mutation might provoke
very particular modifications in the organization of the chromosomes and their environment, so as to sensitize the microtubule spindle to depolymerizing agents. Finally, our data also suggest that the Bmh1/2 pathway activated by cdc13-1 damage is independent from the PKA pathway and its effector, the APC activator, Cdc20.35
Bmh1 may act by decreasing the level of cdc13-1 damage in an Exo1-independent manner. Although this is unlikely, given full Rad53 activation seen at 34°C in the absence of Bmh1 (Fig. S1), we cannot rule out the possibility that a slight decrease in damage at 29°C is sufficient to sustain proliferation. Alternatively, Bmh1 might truly function as a DNA damage checkpoint, as evidenced by the data of Figures 2D and 4C. The bmh1 S189P mutant was hypersensitive to MMS as well as to benomyl, thus suggesting that Bmh1/2 contribute in parallel to consolidation of both the DNA damage and spindle checkpoints. We propose that 14-3-3 proteins, with their large panel of interactions with other mitotic proteins, may be ideally suited to provide more robustness to both types of
mitotic checkpoints. Future work will aim at elucidating the exact molecular mechanism by which the BMH proteins can achieve this control. Studying similar functions of 14-3-3 proteins that are likely to be present in other organisms will represent a major issue in our understanding of uncontrolled proliferation in cancer cells.
Materials and Methods
Strains and plasmids, as well as the methods used for PCR mutagenesis, cell synchronization, cell cycle analysis and immuno-fluorescence are described in the Supplementary material.
Acknowledgements
We thank Andrew Murray, Andrew Emili, Akira Matsuura, Leland Hartwell, Errol Friedberg, Gerald Fink, Wolf-Dietrich Heyer and Giovanna Lucchini for the gifts of strains and plasmids. This work was supported by grants from the “Association pour la Recherche contre le Cancer” and the “Comité Départemental de la Savoie de la Ligue Nationale contre le Cancer”.
Note
Supplementary materials can be found at:
www.landesbioscience.com/supplement/GrandinCC7-17-Sup.pdf
References
1. Aitken A. Functional specificity in 14-3-3 isoform interactions through dimer formation and phosphorylation. Chromosome location of mammalian isoforms and variants. Plant Mol Biol 2002; 50:993-1010.
2. Clémenson C, Marsolier-Kergoat MC. The spindle assembly checkpoint regulates the phosphorylation state of a subset of DNA checkpoint proteins in Saccharomyces cerevisiae. Mol Cell Biol 2006; 26:9149-61.
3. Clerici M, Baldo V, Mantiero D, Lottersberger F, Lucchini G, Longhese MP. A Tel1/MRX-dependent checkpoint inhibits the metaphase-to-anaphase transition after UV irradiation in the absence of Mec1. Mol Cell Biol 2004; 24:10126-44.
4. Downey M, Houlsworth R, Maringele L, Rollie A, Brehme M, Galicia S, Guillard S, Partington M, Zubko MK, Krogan NJ, et al. A genome-wide screen identifies the evolu-tionarily conserved KEOPS complex as a telomere regulator. Cell 2006; 124:1155-68. 5. Fang Y, Liu T, Wang X, Yang YM, Deng H, Kunicki J, Traganos F, Darzynkiewicz Z, Lu L,
Dai W. BubR1 is involved in regulation of DNA damage responses. Oncogene 2006; 25:3598-605.
6. Fesquet D, Fitzpatrick PJ, Johnson AL, Kramer KM, Toyn JH, Johnston LH. A Bub2p-dependent spindle checkpoint pathway regulates the Dbf2p kinase in budding yeast. EMBO J 1999; 18:2424-34.
7. Garber PM, Rine J. Overlapping roles of the spindle assembly and DNA damage check-points in the cell cycle response to altered chromosomes in Saccharomyces cerevisiae. Genetics 2002; 161:521-34.
8. Gardner RD, Burke DJ. The spindle checkpoint: two transitions, two pathways. Trends Cell Biol 2000; 10:154-8.
9. Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol 1995; 15:6128-38.
10. Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006; 440:631-6.
11. Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by system-atic analysis of protein complexes. Nature 2002; 415:141-7.
12. Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O’Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature 2003; 425:737-41. 13. Grandin N, Charbonneau M. Protection against chromosome degradation at the telomeres.
Biochimie 2008; 90:41-59.
14. Grandin N, Damon C, Charbonneau M. Cdc13 prevents telomere uncapping and Rad50-dependent homologous recombination. EMBO J 2001; 20:6127-39.
15. Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 2006; 40:209-35.
16. Hermeking H. The 14-3-3 cancer connection. Nat Rev Cancer 2003; 3:931-43. 17. Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K,
Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002; 415:180-3.
18. Hu F, Wang Y, Liu D, Li Y, Qin J, Elledge SJ. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 2001; 107:655-65.
©2008 LANDES BIOSCIENCE
. DO NO
T DISTRIBUTE
.
19. Jullien D, Vagnarelli P, Earnshaw WC, Adachi Y. Kinetochore localisation of the DNAdamage response component 53BP1 during mitosis. J Cell Sci 2002; 115:71-9. 20. Kakiuchi K, Yamauchi Y, Taoka M, Igawo M, Fujita T, Ito T, Song S, Sakai A, Isobe T,
Ichimura T. Proteomic analysis of in vivo 14-3-3 interactions in the yeast Saccharomyces
cerevisiae. Biochem 2007; 46:7781-92.
21. Krishnan V, Nirantar S, Crasta K, Cheng AY, Surana U. DNA replication checkpoint pre-vents precocious chromosome segregation by regulating spindle behavior. Mol Cell 2004; 16:687-700.
22. Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al. Global landscape of protein complexes in the yeast Saccharomyces
cerevi-siae. Nature 2006; 440:637-43.
23. Lottersberger F, Panza A, Lucchini G, Longhese MP. Functional and physical interactions between yeast 14-3-3 proteins, acetyltransferases, and deacetylases in response to DNA replication perturbations. Mol Cell Biol 2007; 27:3266-81.
24. Lottersberger F, Panza A, Lucchini G, Piatti S, Longhese MP. The Saccharomyces cerevisiae 14-3-3 proteins are required for the G1/S transition, actin cytoskeleton organization and cell wall integrity. Genetics 2006; 173:661-75.
25. Lottersberger F, Rubert F, Baldo V, Lucchini G, Longhese MP. Functions of Saccharomyces
cerevisiae 14-3-3 proteins in response to DNA damage and to DNA replication stress.
Genetics 2003; 165:1717-32.
26. Mackintosh C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regu-late diverse cellular processes. Biochem J 2004; 381:329-42.
27. Mapelli M, Musacchio A. MAD contorsions: conformational dimerization boosts spindle chekpoint signaling. Curr Opin Struct Biol 2007; 17:716-25.
28. Maringele L, Lydall D. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Δ mutants. Genes Dev 2002; 16:1919-33.
29. Pangilinan F, Spencer F. Abnormal kinetochore structure activates the spindle assembly chekpoint in budding yeast. Mol Biol Cell 1996; 7:1195-208.
30. Pinsky BA, Biggins S. The spindle chcekpoint: tension versus attachment. Trends Cell Biol 2005; 15:486-93.
31. Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 1999; 4:153-66.
32. Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297:547-51.
33. Royou A, Macias H, Sullivan W. The Drosophila Grp/Chk1 DNA damage checkpoint controls entry into anaphase. Curr Biol 2005; 15:334-9.
34. Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by Chk1 and Rad53 protein kinases through distinct mechanisms. Science 1999; 286:1166-71.
35. Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nat Cell Biol 2004; 6:138-45.
36. Stern BM, Murray AW. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr Biol 2001; 11:1462-67.
37. Takada S, Kelkar A, Theurkauf WE. Drosophila chekpoint kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell 2003; 113:87-99.
38. Tan ALC, Rida PCG, Surana U. Essential tension and constructive destruction: the spindle checkpoint and its regulatory links with mitotic exit. Biochem J 2005; 386:1-13. 39. Usui T, Petrini HJ. The Saccharomyces cerevisiae 14-3-3 proteins Bmh1 and Bmh2 directly
influence the DNA damage-dependent functions of Rad53. Proc Natl Acad Sci USA 2007; 104:2797-802.
40. van Hemert MJ, Steensma HY, van Heusden GPH. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. BioEssays 2001; 23:936-46.
41. van Heusden GPH, Steensma HY. 14-3-3 proteins are essential for regulation of RTG3-dependent transcription in Saccharomyces cerevisiae. Yeast 2001; 18:1479-91.
42. van Heusden GPH, Steensma HY. Yeast 14-3-3 proteins. Yeast 2006; 23:159-71. 43. Wang Y, Hu F, Elledge SJ. The Bfa1/Bub2 GAP complex comprises a universal checkpoint
required to prevent mitotic exit. Curr Biol 2000; 10:1379-82.
44. Wang H, Liu D, Wang Y, Qin J, Elledge SJ. Pds1 phosphorylation in response to DNA dam-age is essential for its DNA damdam-age checkpoint function. Genes Dev 2001; 15:1361-72. 45. Weinert TA, Hartwell LH. Cell cycle arrest of cdc mutants and specificity of the RAD9
checkpoint. Genetics 1993; 134:63-80.
46. Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 1994; 8:652-65. 47. Wilker E, Yaffe MB. 14-3-3 proteins-a focus on cancer on human disease. J Mol Cell