Bootstrap Embedding for Molecules
Texte intégral
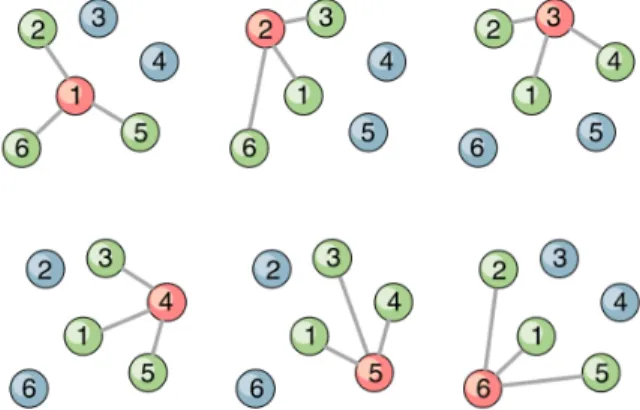
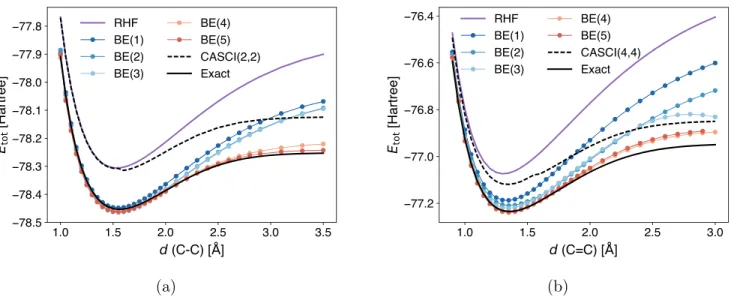
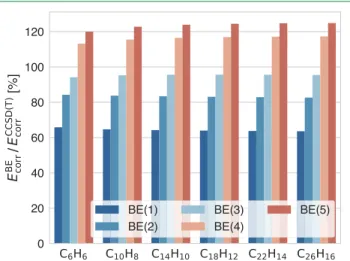
Figure

Documents relatifs
Synthesis, Characterization, and Biochemical Evaluation of Novel Heterocyclic Stilbene Compounds with Tubulin Targeting
It was shown in the previous work that α pseudopo- tentials could accurately reproduce absorption spectra for the excitations of the remaining electrons in the sys- tems (as
Abbreviations: APA, antiproliferative assay; AVA, antiviral assay; C+, positive control; C-, negative control; CC, compound control; CWT, cells without treatment; DMSO,
Abstract - The Mossbauer Effect in 5 7 ~ e is used to discover and characterize chemical species formed between bare iron atoms or molecules with molecules of quasi-inert gases
A newly proposed mixed-element de- embedding technique that removes the contact resistance, the pad capacitance and a transmission line segment from the IUT yields better results
The paper is organized as follow. In Section 2, we provide a general notation, basic facts on real analyticity, and a result on elliptic regularity, that was essentially present in
3.1 the general problem of constructing Hamiltonian integrable perturbations of dispersionless systems, we will exploit the connection of the Principal Hierarchy of X ad 1 with a
-- la difficulté à instruire les dossiers de demandes de chômage partiel des grandes entreprises, comme l’automobile par exemple (le niveau de décision, comme le niveau
