1
Plan
Introduction ………6
I- Définition et données générales………7
II- Objectifs………...8
III- Rappel sur les facteurs V et VIII………...8
1- Structure………...8 2- Gène………10 3- Lieu de synthèse………...12 4- Propriétés………....12 5- Activation………....13 6- Rôle……….16 7- Inactivation……….18
2
Première partie : le déficit combiné en facteur V et VIII de
la coagulation dans la littérature………..20
1
erchapitre : syndrome hémorragique par déficit combiné
en facteur V et VIII………...21
I- Historique………22
1- Historique, Le mystère du déficit combiné en FV et VIII……23
2- Les hypothèses physiopathologiques du déficit combiné…….23
a- Hypothèse du précurseur commun………....23
b- Hypothèse d’un déficit en protéine C………23
c- Hypothèse d’une sulfatation défectueuse de la tyrosine des facteurs V et VIII………...24
3- La découverte de la première protéine impliquée dans ce déficit combiné……….24
4- La découverte de la deuxième protéine impliquée dans ce déficit combiné………..
II- Epidémiologie………..26
1- Fréquence du déficit combiné………...26
2- Répartition géographique………26
III- Aspect moléculaire du déficit combiné………...27
3
2- Structure des protéines LMAN1 et MCFD2………27
a- LMAN1………27 b- MCFD2……….28 3- Rôle de LMAN1 et de MCFD2………..31 4- Mutations de LMAN1 et de MCFD2………31
IV- Physiopathologie………...34
V- Transmission génétique……….35
VI- Manifestations cliniques………...36
1- Hémorragies spontanées ou provoquée par un traumatisme minime………..37
a- Hémorragies extériorisées……….37
a1- Hémorragies de la peau………..37
a2- Hémorragies des muqueuses………..37
b- Hémorragies non extériorisées………...37
2- Hémorragies provoquées par un acte chirurgical……….37
VII- Diagnostique biologique……….38
VIII- Traitement et prise en charge………..39
1- Traitement du déficit combiné en facteur V et VIII…………39
4
b- La desmopressine………..39
c- Le concentré de facteur VIII………..40
d- Schéma thérapeutique………40
2- Prise en charge de la femme enceinte et de son bébé…………42
a- La femme enceinte……….42
b- Le nouveau né………43
2
emechapitre : une nouvelle approche de la synthèse et la
sécrétion des facteurs V et VIII………...44
I- Maturation des facteurs V et VIII………..44
1- Au niveau du réticulum endoplasmique……….44
2- au niveau du compartiment intermédiaire……….46
3- Au niveau de l’Appareil de Golgi………...47
II- sécrétion des facteurs V et VIII hors des cellules………...48
III- Facteurs V et VIII dans la circulation……….48
Deuxième partie : Notre observation………51
I- Antécédents et histoire familiale………...52
II- Expression clinique……….52
5
Conclusion générale……….54
Résumé………..56
6
7
I-
Définition et données générales:
Le déficit combiné en facteurs V et VIII de la coagulation est un désordre héréditaire, qui représente la forme la plus fréquente d’anomalie constitutionnelle, associant plus d’un facteur de coagulation, et n’étant pas lié à la coïncidence accidentelle de plusieurs déficits héréditaires génétiquement distincts, mais lié à une anomalie génétique unique1.
En effet, le nombre de familles dans lesquelles a été observé ce déficit associé, est très supérieur au nombre théorique qu’on pourrait attendre d’une association due au hasard.
Dans le domaine de la coagulation, un déficit en protéine de la coagulation était pour l’instant lié à une anomalie du gène producteur de cette protéine. Pour la première fois, le déficit est lié à une anomalie de sécrétion de plusieurs facteurs de la coagulation.
Les patients affectés présentent une hémophilie modérée, en rapport avec le niveau plasmatique de ces deux facteurs allant de 5 à 30% des taux normal2, 3(5 à 3OU/dl), aussi bien en activité qu’en antigène, dont la symptomatologie est similaire à un simple déficit en facteur VIII (hémophilie A mineure) ou en facteur V.
Il se transmet sous le mode autosomique récessif.
Le traitement de ce déficit combiné dépend de la nature de saignement et du niveau plasmatique des facteurs V et VIII.
8 II- Objectifs
Nos objectifs seront donc de :
Faire connaître cette maladie « rare », en rapportant des données très récentes de la littérature. Cette connaissance est rattachée à celle de la sécrétion des facteurs V et VIII et l’intérêt que peut présenter cette sécrétion dans le domaine thérapeutique.
Rapporter le premier cas marocain de déficit combiné en FV/FVIII le diagnostic est réalisé au service d’Hématologie Biologique de CHU-Rabat.
III- Rappels sur les facteurs V et VIII
Le facteur V (pro accélérine) et le facteur VIII (anti hémophilique A) font partie, avec la cèruloplasmine, d’une famille de molécules caractérisées par des homologies de structure primaire, ces homologies sont particulièrement importantes pour les facteurs V et VIII, qui ont par ailleurs une fonction comparable(1).
1. Structure des F V et VIII:
Ces deux protéines ont une structure très voisine (17, 18, 19) : Le facteur V (FV) et le facteur VIII (FVIII) sont synthétisés sous forme de polypeptide à chaîne unique, respectivement de 2196(10, 16) et de 2332 (12) acides aminés (aa), après libération du peptide signal (28 aa (15, 16) pour le facteur V et 19(12) pour le facteur VIII).
9 Concernant le facteur VIII, dés qu’il est synthétisé il est clivé en une chaîne d’hétéro dimère.
De l’extrémité N-terminale à l’extrémité C-terminale, les domaines s’enchaînent ainsi : A1, A2, B, A3, C1, C2 (10, 15, 16)
, avec des zones hyper acides (14), situées entre les domaines A1, A2 et A2, B d’une part et entre B, A3 d’autre part, pour le facteur VIII ; et entre les domaines A2, B d’une part, et entre B, A3 d’autre part pour le FV.
Le domaine A de ces deux protéines, présente une homologie de 40%, et présente une homologie de 30% avec le domaine A de la cèruloplasmine
(transporteur de cuivre) (10, 15), suggérant un rôle dans la liaison métal-ion(8). Il est constitué de la réplication de trois séquences similaires : A1 (1-303),
A2 (317-656) et A3 (1546-1877) (10) pour le facteur V et A1 (1-336), A2 (373-710) et A3 (1690-2019) (20) pour le facteur VIII.
Le domaine B des deux facteurs a une structure complexe et unique, et présente une homologie de 15% (15).
Pour le facteur V : B (710-1545), est entièrement codé par l’exon 13, il contient 2 répétitions de 17 aa, et 31 répétitions de 9 aa qui sont absents dans le domaine B du facteur VIII (15, 16), en plus de 25 sites potentiels à la N- glycosylation (15, 18).
Pour le facteur VIII : B (741-1648) (19), codé par l’exon 14 et contient 19 sites pour la N- glycosylation (17, 18)
A la différence des domaines A et C, ce domaine ne participe pas à la coagulation.
10 Le domaine C : de ces deux protéines, présente une homologie de 40% à 46%. Il est constitué par la répétition de deux séquences ;
Pour le facteur V : C1 (1878-2036) et C2 (2037-2196)10. Pour le facteur VIII : C1 (2020-2172) et C2 (2173-2332) (20).
Ce domaine comporte des sites de fixation aux phospholipides au niveau C terminal (19, 20), en plus d’un site de fixation au facteur de Von Willebrand par l’extrémité N terminale pour le facteur VIII, mais, il ne fixe qu’à l’un des deux, selon son état d’activation(9)
.
2. Gène des facteurs V et VIII:
Le gène du FV a approximativement une taille de 80kilo bases (kb), localisé sur le bras long du chromosome 1 (1q23) composé de 24 introns et 25 exons (15,23).
Ce gène est transcrit en ARN messager (ARNm) de 6,8kb(10,15) qui est traduit en un précurseur, le pro peptide (10) de 330KD, contenant 2224aa(15) (figure 2).
Celui du FVIII est un long gène s’étendant sur 186 (kb) (20), situé sur l’extrémité terminale du bras long du chromosome X (Xq28) (20)
comportant 25 introns et 26 exons(43, 46).
Les 26 exons codent un ARNm de 9kb (20) qui est lui même traduit en une protéine de 280KD, composée de 2351aa (20) (figure 1).
11 Figure 1 : le facteur VIII, du gène (A) avec ses 26 exons à la protéine (B) avec ses trois domaines (A, B, C) 12.
Figure 1 : le facteur V du gène (A’) avec ses 25 exons, à la protéine (B’) avec ses trois domaines (A, B, C) 19.
C1 A1 A2 B A3 C2 Peptide signal B A1 A2 B A3 C1 C2 Peptide signal B’ A’
12
3. Lieu de synthèse des facteurs V et VIII
Le FVIII a principalement une origine hépatique mais également extra hépatique (tableau2).
Le facteur V plasmatique est synthétisé dans l’hépatocyte et dans la cellule endothéliale, le facteur V plaquettaire dans le mégacaryocyte.
FVIII
Hépatocytes mais aussi par le rein, la rate, les ganglions lymphatiques, le muscle et le placenta (12).
FV-Plasmatique
FV-Plaquettaire
Dans l’hépatocyte (1, 15)
, et dans la cellule endothéliale vasculaire (1, 10).
Dans le mégacaryocyte (1, 10, 15).
Tableau 1: lieu de synthèse des F V et VIII
4. Propriétés des facteurs V et VIII (14):
Le F V est présent dans le plasma à un taux 40 fois plus élevé que celui du FVIII avec une demi-vie plus longue.
Certaines caractéristiques des F V et VIII sont résumées dans le tableau2.
Concentration plasmatique µg/ml Demi-vie biologique (h)
FV 5-10 12-36
FVIII 0.1-0.2 10-12
13
5. Activation des facteurs V et VIII:
Ces deux facteurs circulent sous forme inactive, monomérique pour le FV et hétérodimérique pour le FVIII.
Leur activation est produite essentiellement par la thrombine et accessoirement par le facteur Xa(1).
Pour le facteur V : le clivage se fait à trois niveaux, éliminant deux polypeptides centraux composant le domaine B, et qui sont fortement glycosylés, de 150 et 71KD (710-1018aa et 1019-1545 aa respectivement) (10), suggérant qu’effectivement le domaine B n’a aucun rôle dans la coagulation.
La thrombine agit d’abord sur l’arginine 709, et puis sur l’arginine 1018 et 1545 produisant un heterodimère qui constitue le FV activé ou l’accélérine, formé d’une chaine lourde de 94KD (A1,A2 :1-713 aa), associée à une chaine légère de 71 ou de 74KD (A3-C1-C2 :1537-2183 aa) par des liaisons non covalentes calcium dépendantes(10, 19), cette différence de poids moléculaire de la chaine légère est due à la présence de deux formes(iso formes) ; FV1=74KD, FV2=71KD ; résultant de la différence de la glycosylation au niveau du domaine C2(16).
Ces deux formes diffèrent par leurs fonctions (10,15) :
Liaison aux phospholipides : V1 se lie moins bien aux phospholipides anioniques que V2.
14 V1 produit plus de thrombine que V2.
Pour le facteur VIII : La thrombine agit d’abord sur la chaine lourde, au niveau de l’arginine 740 et 372 pour produire deux polypeptides de 43KD (A2) et de 50KD (globalement A1), et au niveau de la chaine légère en arginine 1689 pour donner un monomère de 73 KD (A3, C1, C2) (12, 18) . Le clivage au niveau de ces sites entraine une élimination du domaine B avec réalisation d’une liaison entre A1et A3 par ion de calcium(9)
15
VIII
V
Figure 2 : structure schématique des facteurs VIII et V (10, 19). H2 IIa(1018) 300KD A1 A2 B A3 C1 C2 NH2 NH2 COOH COOH F V Circulant 94KD IIa(709) 709 73KD Ca2+ IIa(1545) 71KD 150KD NH2 COOH F V Activé 55KD 50KD 43KD 73KD
IIa( 372) IIa( 740) IIa(1689)
Calcium
NH2 COOH F VIII Activé
A3 C1 C2 A1 A2 B NH2 IIa(1313) IIa(1648) Calcium COOH F VIII Circulant Chaine lourde Chaîne légère
80KD 200KD
A1 A2 B A3 C1 C2 NH2
16
6. Rôle des facteurs V et VIII dans la cascade de la coagulation
Ces deux protéines jouent un rôle crucial dans la cascade de coagulation. Elles interviennent comme des cofacteurs, qui accélèrent considérablement les réactions enzymatiques des sérines protéases.
Le FV accélère l’activation de la prothrombine (II) en thrombine, par le facteur Xa (7, 8, 9) (figure 4), grâce à des interactions protéine / protéine/ surface lipidique; en effet le FV n’agit qu’à la surface des phospholipides auxquelles il est lié par des liaisons hydrophobes. Il est ainsi contigu au facteur X et à la prothrombine. La prothrombine présente également au niveau de son domaine 2 une partie du site de fixation pour le FV (8, 10, 11).
Concernant le FVIII, il agit dans la voie intrinsèque de la coagulation et à son tour il accélère l’activation du facteur X par le facteur IX en présence d’ion calcium et de phospholipides (12, 13) (figure 4).
17 Voie intrinsèque voie extrinsèque
In vitro: verre, kaolin, célite…
XII XIIa XI XIa IX IXa X VIIIa PL Ca++ Xa II Va PL Ca++ I IIa Ia (monomère de fibrine qui sera stabilisé par une
transglutaminase le FXIIIa).
Figure 3: Rôle des facteurs V et VIII dans la cascade de la coagulation.
VII VIIa [FT, PL, Ca++]
18
7. Inactivation des facteurs V et VIII:
Ces deux protéines subissent l’action protéolytique de la protéine C, activée elle-même par la thrombine en présence de la thrombomoduline (cofacteur vasculaire).
Le clivage du FV se fait au niveau de la chaîne lourde, au niveau de l’arginine 506, l’arginine 306 et l’arginine 679 suivant cet ordre, mais ce dernier clivage en arginine 679 est le plus lent et n’apparaît pas important pour l’inactivation du FV, produisant ainsi le dégagement spontané du domaine A2, et la perte complète de l’activité du FV (10, 15)
(figure 5). Le clivage du FVIII se fait au niveau de la chaîne lourde, au niveau des domaines A1 et A2 en arginine 336 produisant une forme inactive constituée du domaine A1 (1-336) de 45KD associé à la chaine légère (A3-C1-C2) de 67KD (20) (figure 5).
19 . F V activé FV inactivé
FVIII activé FVIII inactivé
Figure 4: Structures schématiques des F V et VIII activé et inactivé19. 67KD 45KD A1 A2 A3 C1 C2 Ca++ 336 A1 C1 C2 PCa, IIa, Xa Xa Ca2+ A1 A2 A3 C1 C2 A1 A3 C1 C2 A2 C A2 NN N 306 506 Ca2+ PCa+PS
20
1ère partie :
Le
déficit combiné
en facteur V et VIII
dans la littérature
21
1èr chapitre :
Syndrome hémorragique
par déficit combiné en
22 I- Historique :
1- Historique, le mystère du déficit combiné en facteur V et VIII :
En 1954 : ce déficit combiné en FV et VIII a été décrit pour la première fois chez une famille par Oeri et al.
En 1958 : ce déficit a été décrit chez quatre membres de trois familles, suggérant que cette anomalie n’étant pas liée à la coïncidence de deux déficits héréditaires génétiquement distincts (hémophilie et parahémophilie) (24).
Jusqu’au 1969 : on note cinq familles parmi huit soufrant de ce déficit, descendants tous de consanguinité parentale (25).
En 1972 : Smit Sibinga et al arrivent à conclure qu’il s’agit d’une maladie autosomale récessive, avec une expression variable chez les hétérozygotes, et ceci après de longues études sur de nombreuses familles (24).
Comment expliquer le mécanisme de ce désordre héréditaire de deux gènes de localisation distincte, l’un sur le chromosome X et l’autre sur le chromosome 1 ?
Les chercheurs se sont tournés vers la génétique moléculaire pour expliquer cette énigme (25).
23
2- Les hypothèses physiopathologiques du déficit combiné :
a- L’hypothèse du précurseur commun des FV et VIII:
Dans les années 1967, vu la grande homologie entre les FV et FVIII, une équipe pose l’hypothèse d’un problème au niveau de la synthèse d’un précurseur commun des FV et VIII de la coagulation.
Cette hypothèse a été testée plus tard, mais vite écartée (25).
Figure 5 :l’hypothèse du précurseur commun des facteurs V et VIII25 b- L’hypothèse d’un déficit en inhibiteur de la protéine C :
Dans les années 1980 : Marlar et Griffin posent l’hypothèse d’un déficit en inhibiteur de la protéine C, impliquant une augmentation de la protéine C et donc une inactivation des FV et VIII.
Par la suite, il a été montré que cette hypothèse était liée à des artefacts de laboratoire vue l’instabilité de la protéine C vis-à-vis de la congélation répétée et la fusion, alors qu’aucun déficit en inhibiteur de la protéine C n’a été détecté(25)
.
Précurseur
Facteur V
24 c- Hypothèse d’une sulfatation défectueuse des tyrosines des facteurs V
et VIII :
En1992 : Debra Pittman et Kaufman Lascif ont proposé l’hypothèse d’une sulfatation anormale des tyrosines, pouvant être responsable de ce déficit combiné, sachant l’importance de cette sulfatation dans la maturation de ces deux facteurs.
Cependant, les résultats étaient décevants, aucune sulfatation défectueuse de tyrosine des FV et VIII n’a été retrouvée (25).
En vue de découvrir le mystère de cette maladie, il y a eu des collaborations entre un grand nombre de chercheurs, essayant de localiser le gène responsable, à l’aide d’une étude par clonage positionnel, et qui était à l’époque une technique relativement nouvelle.
3- La découverte de la première protéine impliquée dans ce déficit combiné :
Après de longues études, une équipe de l’université de Michigan, avait localisé, en 1997, le locus responsable de ce déficit combiné sur le bras long du chromosome 18, entre les marqueurs D18S849 et D18S64.
La découverte de ce locus, a permis d’exclure l’intervention de toute protéine de l’hémostase, puisqu’aucune d’entre elle n’a le gène au niveau de ce chromosome, suggérant qu’il s’agit plutôt d’une anomalie au niveau d’une voie commune de biosynthèse des FV et VIII.
25 En effet, cette même équipe a pu identifier le gène responsable par clonage positionnel.
En 1998, Nichols et al ont décrit deux mutations fondatrices distinctes qui conduisent à l’absence totale du produit du gène en question.
Ce gène code pour une protéine cytoplasmique, identifiée depuis 1987, comme un marqueur du compartiment intermédiaire entre le Reticulum Endoplasmique et l’Appareil de Golgi, appelée ERGIC53 (Endoplasmique Réticulum Golgi Intermédiaire Compartiment 53) actuellement appelée LMAN1 pour lectin mannose binding 1(25, 41).
L’hypothèse selon laquelle cette protéine, LMAN1, jouait le rôle d’une protéine chaperonne pour le transport des glycoprotéines sécrétées au niveau des compartiments intracellulaires avait alors été proposé (24)
A partir de 1999, de nouvelles mutations commençaient à apparaitre, toujours au niveau de cette protéine (24).
En revanche, chez approximativement 30% des familles souffrant de ce déficit, aucune anomalie n’a été détectée dans le gène LMAN1, suggérant que cette protéine n’est pas la seule impliquée dans ce déficit combiné(25).
4- La découverte de la deuxième protéine impliquée dans ce déficit combiné :
En 2003 : Zhang et al ont découvert un deuxième gène, localisé sur le bras court du chromosome 2, et codant pour une protéine appelée
26 MCFD2 pour Multiple Coagulation Factor Deficiency 2, responsable de ce déficit combiné chez les familles sans mutation sur LMAN1(24). De nombreuses mutations au niveau de ce gène (25) ont été découvertes.
Il persiste toujours des cas de ce déficit combiné qui ne sont pas liés au désordre de ces deux gènes LMAN1 et MCFD2 suggérant fortement un troisième locus impliqué dans ce déficit.
II- Epidémiologie :
1- Prévalence du déficit combiné en facteur V et VIII :
Le déficit combiné en facteur V et VIII est très rare, sa fréquence est estimée entre 1 /100000 et 1/1000000(26).
On note une prévalence plus importante chez les populations à forte consanguinité (1/100000(39) comme celle des juifs Séfarades du moyen orient, population très étudiée depuis de nombreuses années (27, 28, 39).
2- Répartition géographique :
Une centaine de familles souffrant de ce déficit a été décrite dans le monde à ce jour, la majorité de ces cas se retrouvent autour du bassin méditerranéen : Israël, Iran, Italie ; avec une partie en Amérique du Nord Amérique du Sud, au Japon et en Inde (27, 45).
27 III- Aspect moléculaire du déficit combiné en F V et VIII
1- Gènes de LMAN1 et de MCFD2
Le locus responsable de ce déficit combiné avait été localisé en 1997 sur le bras long du chromosome 18 (18q21). Il contient 13 exons et 12 introns, et code pour une protéine appelée ERGIC-53 ou LMAN12,32.
En 2003, un deuxième gène a été isolé chez des patients sans mutation du gène LMAN1, il est situé sur le bras court du chromosome 2 (2p21), et contient 4 exons et s’étend sur 19kb. Ce gène code pour une protéine nommée Multiple Coagulation Factor Deficiency 2 (MCFD2)2, 5 .
2- Structure des protéines LMAN1 et MCFD2 :
a- LMAN1:
Est une protéine transmembranaire non glycosylée, décrite comme une lectine spécifique des résidus mannoses, appartenant à une nouvelle classe de lectines intracellulaires dont la structure est proche de celle des lectines des légumineuses, composée de 499 à 51O aa avec un poids moléculaire de 53KD(40).
LMAN1 se présente sous forme d’un hexamère comportant plusieurs régions importantes pour sa fonction :
CD : Cytosolic Domain ; très conservé, contenant un motif (KKFF) (507 lys lys Phe Phe510).
28
SS : Séquence Signal de 30 aa33.
TMD : Transmembran Domain ; contenant 18 aa et contribuant à la rétention dans le Réticulum Endoplasmique33.
Stalk : contient de nombreux résidus cystéines, surtout C466 et C475, nécessaires à la polymérisation de cette protéine en dimère ou en hexamère (42).
CRD : Carbohydrate Recognition Domain ; qui sert à la reconnaissance et à la fixation des groupements mannoses des glycoprotéines au niveau luminal(44).
b- MCFD2:
Est une protéine luminale soluble non glycosylée de 145 aa, et d’un poids moléculaire de 16 KD3, 35. MCFD2 possède :
Un peptide signal dans son extrémité N terminal.
Deux domaines EF hands, dans son extrémité C terminal, contenant un site potentiel de fixation protéine-protéine calcium dépendante, supposant une association possible, dans un complexe, avec LMAN13,35 .
29
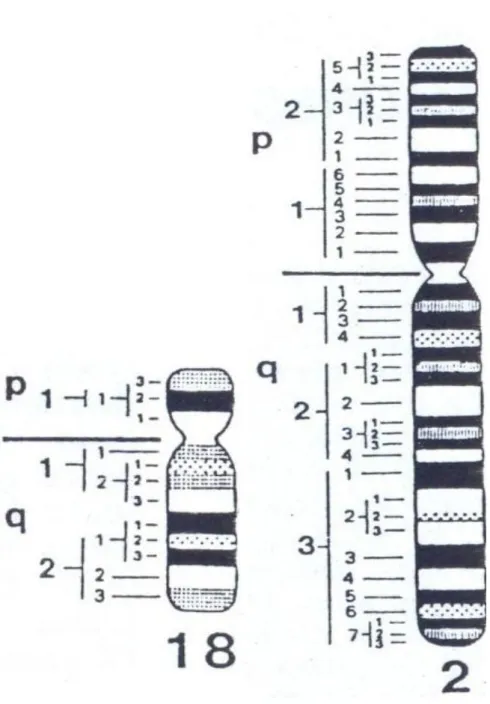
Figure 6 : les différentes régions des chromosomes 2 et 18(6).
Le gène de LMAN1 est situé sur la région 21.3 du bras long du chromosome 18.
30 LMAN1
MCFD2
Figure 7 : LMAN1 et MCFD2 : du gène à la protéine.
A, A’ : les gènes respectivement de LMAN1 et de MCFD2 avec leurs 13 et 4 exons.
B, B’ : les protéines respectivement de LMAN1 et de MCFD2. KKFF Signal CRD Stalk TMD CD A B 1 13 5’ 1 2 3 4
Signal EF-1 EF-2 A’
31
3- Le rôle de LMAN1 et de MCFD2 :
Etant donné leur localisation intracellulaire, dans le compartiment intermédiaire de Golgi, le complexe LMAN1-MCFD2 mannose et calcium dépendant semble jouer un rôle dans le transport des glycoprotéines (FV, FVIII) sécrétées au niveau des compartiments intracellulaires5, 36.
LMAN1 serait indispensable à l’échange du complexe COatomére Protéine II (COPII) pour COatomére Protèine I (COPI) au moment où se fait le choix entre un transport antégrade (réticulum endoplasmique vers golgi) et rétrograde (Golgi vers réticulum endoplasmique).
MCFD2 n’est pas indispensable à la sortie et au recyclage des hexamères de LMAN1, mais plutôt, MCFD2 servirait de cofacteur pour la recapture des glycoprotéines correctement conformées et glycosylées 26, et pourrait être impliqué dans le phénomène de relargage de ces glycoprotéines dans l’Appareil de Golgi.
4- Les mutations de LMAN1 et de MCFD2 :
Les anomalies responsables du déficit combiné en facteur V et VIII peuvent toucher n’importe quelle partie du gène.
On note l’identification de 30 mutations du gène LMAN1 et 9 du gène MCFD2 3,36.
32 Ces mutations peuvent être classées en 5 catégories :
Mutations non sens : avec 6 mutations au niveau de LMAN1.
Délétions et insertions : induisant un décalage du cadre de lecture (DCL), avec 13 pour LMAN1 et 3 pour MCFD2.
Une mutation sur le premier codon Met notée pour LMAN1.
Anomalie d’épissage : on note 8 pour le gène LMAN1et 3 pour MCFD2.
Mutations faux sens : 2 pour LMAN1 et 3 pour MCFD2.
En plus de la découverte de 2 nouvelles mutations au niveau du gène MCFD2 ce qui lui donne un total de 11 mutations3.
Tableau 3 : mutations décrites dans le gène de LMAN130, 38
Localisation Mutations Conséquence Origine géographique Exon1 Met 1 Thr Mutation sur le premier Italie
33 codon Met
Exon 1 Del G 23 DCL Iran
Exon 1 Del G 31 DCL Algérie
Exon 1 Ins G 89 DCL Iran
Exon 1 Ins G 85 DCL Venezuela
Exon 2 Gly 114 Stop Non sens Inde
Exon 3 Del C 422 DCL Japon
Exon 5 Arg 202 Stop Non sens Japon, Iran Intron 5 IVS G 5+1 T Anomalie d’épissage Italie
Exon 6 Del 16 pb 720 -735 DCL Venezuela
Exon 7 Del 781 T DCL Australie
Exon 7 G 822 A Anomalie d’épissage Iran Intron 7 IVS G 7+1 A Anomalie d’épissage Belgique Intron 7 IVS G 7+33 ins
GGTT
Anomalie d’épissage Etats- unis Intron 7 IVS G 7-1 C Anomalie d’épissage Thaïlande
Exon 8 Del A 841 DCL Pologne, Pakistan
Exon 8 Lys 302 Stop Non sens France
Exon 8 Ins A 912 DCL Iran
Exon 9 Glu 321 Stop Non sens Iraq
Exon 9 Del TC 1109-1112 DCL Etats- unis
Exon 9 Gln 380 Stop Non sens Liban
Intron 9 IVS T 9+2 G Anomalie d’épissage Iran Intron 9 IVS T 9+2 C Anomalie d’épissage Tunisie
Exon 10 Ins T 1208-1209 DCL Italie
Exon 10 Del 5 pb 1214-1219 DCL Iran
Exon 11 Arg 456 Stop Non sens Chine, Pakistan, Thaïlande. Exon 11 Del G 1271 anomalie d’épissage Liban
Exon 12 Cys 475 Arg Faux sens Argentine d’origine Italienne Exon 12 Cys 475 Ala Faux sens
Exon 13 Del A 1524 DCL Etats- unis
34 Localisation Mutations Conséquence Origine
géographique Intron 1 INV G 1-1 C Anomalie d’épissage Iran
Intron 2 INV G 2+5 A Anomalie d’épissage Italie, Inde, Serbie Intron 3 INV G 3+1 A Anomalie d’épissage
Exon 2 Del C 103 DCL Italie
Exon 3 Del T 249 DCL Turquie
Exon 3 Del 8 pb 263-270 DCL Afrique du Sud
Exon 3 Asp89Ala Faux sens
Exon 4 Asp 129 Glu Faux sens Etats-Unis
Exon 4 Ile 136 Thr Faux sens Kosovo,
Venezuela
IV- Physiopathologie:
Ce déficit combiné en F V et VIII se différencie des autres déficits de la coagulation, qui concerne un facteur isolé, comme l’hémophilie A ou B. D’après des études familiales établies, il a été montré récemment que ce déficit est lié à une seule anomalie génétique responsable du double déficit, il s’agit d’une anomalie de production au niveau d’une étape commune de sécrétion de ces deux facteurs.
En effet ce déficit combiné résulte d’un défaut du gène, qui entraine une diminution simultanée de ces deux facteurs, cette anomalie peut être soit liée à la présence d’une protéine anormale ERGIC53/LMAN1, et qui est
35 indispensable au transport de ces deux protéines du Réticulum Endoplasmique vers l’Appareil de Golgi, ou à son absence.
ERGIC53/LMAN1, en cas de son absence, l’autre protéine MCFD2 montre également un taux très faible, suggérant une interaction forte entre ces deux protéines.
Dans ce cas, il n’y aura plus possibilité de transporter les deux facteurs de coagulations V et VIII, et donc une diminution de leur taux par diminution de leur sécrétion, et par conséquent la coagulation du sang sera très diminuée, ce qui va entrainer des manifestations hémorragiques.
V- Transmission génétique :
Le déficit en facteur V et VIII est une maladie rare.
Sa transmission est autosomale, affectant aussi bien les hommes que les femmes, et récessive, avec une pénétrance partielle et expression variable chez les hétérozygotes1, 4, 37.
Dans ce cas les individus atteints sont homozygotes pour une mutation donnée, ou double hétérozygote (37).
Dans certains cas extrêmement rares, ce déficit combiné peut avoir une autre cause ; plutôt de découler d’un seul défaut génétique affectant les FV et VIII au niveau du chromosome 18 ou du chromosome 2, un enfant
36 peut hériter un gène du facteur V défectueux (1q), et un gène du facteur VIII défectueux (Xq28) de ses deux parents.
Les risques d’hériter un déficit en facteur V soit 1 /1000000, et les risques d’hériter un déficit en facteur VIII est estimé à 1/10000.
Par conséquent, le risque d’hériter les deux gènes défectueux séparément est équivaut à 1 sur 10 Milliards.
Pourtant une telle transmission simultanée a été décrite chez quatre familles (45).
On note également un autre type de transmission, concernant une patiente Thaïlandaise, ayant hérité de ses parents deux mutations différentes, dans ce cas on parlait d’une transmission hétérozygote composite (29).
VI- Manifestations cliniques :
Il s’agit d’un syndrome hémorragique variable, dans la sévérité dépend surtout du taux du facteur VIII.
La symptomatologie est similaire à celle d’une hémophilie mineure. Mais, en général, cette symptomatologie hémorragique est assez modeste, voire inexistante et généralement liée à un traumatisme ou une chirurgie, puisque les niveaux circulants des facteurs V et VIII semblent
37 suffisant pour maitriser les saignements spontanés qui sont par ailleurs très rares.
Chez les homozygotes, les hémorragies les plus fréquentes qu’on puisse noter sont:
1- Hémorragies spontanées ou provoquées par un traumatisme minime :
a- Hémorragies extériorisées :
a1- Hémorragies de la peau1, 31, 37 : Des ecchymoses.
Saignements prolongés après coupure.
a2- Hémorragies des muqueuses31, 37, 40 : Des épistaxis.
Des ménorragies Des gingivorragie
b- Hémorragies non extériorisées :
Des hématomes et des hémarthroses sont également observés 37, 40.
2- Hémorragies provoquées par un acte chirurgical :
Ce type d’hémorragie est commun à tous les individus atteints de ce déficit combiné, avec principalement1, 31, 37 :
38 Des saignements après extraction dentaire.
Des saignements après intervention chirurgicale. Des hémorragies du post partum.
VII- Diagnostic biologique :
L’activité anticoagulante des facteurs V et VIII est mesurée à l’aide du temps de Quick (TQ) et du temps de céphaline activée (TCA).
Chez les homozygotes, les examens biologiques du déficit combiné mettent en évidence 1, 34:
Un allongement du TQ. Un allongement du TCA.
Un temps de saignement normal.
Le dosage des facteurs de coagulation montre :
Une concentration plasmatique du facteur V varie de 1 à 33%. Une concentration plasmatique du facteur VIII varie de 1 à 31%. Une concentration des autres facteurs étant normale.
Le diagnostic moléculaire est indispensable pour identifier le désordre précis sur les gènes LMAN1 et MCFD2
39 VIII- Traitement et prise en charge
1- Traitement du déficit combiné en facteur V et VIII
Le traitement de ce déficit combiné dépend de la nature de saignement et du niveau plasmatique des facteurs V et VIII31, 37.
Les traitements existant pour maitriser le saignement : Le plasma frais congelé.
La desmopressine.
Le concentré de facteur VIII.
Plasmatique.
Recombinant.
a- Le plasma frais congelé:
Le plasma normal qui renferme les facteurs V et VIII reste la seule source du FV en absence du FV recombinant ou du concentré de FV, dont l’adjonction corrige les anomalies observées.
b- La desmopressine :
La 8- Déamino -D – Arginine Vasopressine ou la DDAVP, dont la forme injectable est commercialisée sous le nom de MINIRIN*.
C’est un analogue structural de la vasopressine, dérivé de l’adrénaline, qui se différencie de cette hormone naturelle par :
40 - Une augmentation de l’activité antidiurétique.
- Ses effets sur le facteur VIII.
Cette desmopressine induit la libération du facteur de Von Willebrand hors de ses sites de stockage endothélial, et par conséquent l’augmentation de sa concentration, la concentration du FVIII, et celle de l’activateur tissulaire du plasminogène.
Du fait de son effet antidiurétique, il peut exister un risque d’hyponatrémie, en cas d’injection répétée ou chez le petit enfant, dans ces circonstances, il faut imposer une restriction hydrique et une surveillance de la natrémie.
Mais il n’est efficace que s’il existe une synthèse même minime du facteur VIII et du facteur de Von willebrand, ainsi il ne peut être utilisé quand cas du déficit léger en facteur VIII.
c- Le concentré de facteur VIII :
Ce type de concentré ne renferme que le facteur VIII, et n’est envisagé que si l’efficacité du DDAVP, sur le taux de facteur VIII, parait insuffisante.
d- Schema thérapeutique 31 :
Traitement des épisodes hémorragiques spontanés :
Concernant ce cas l’adjonction du plasma normal semble pouvoir corriger ces saignements, sinon il peut être associé au concentré de facteur VIII.
41 Traitement des épisodes hémorragiques mineurs provoqués par un
traumatisme:
Dans ce cas le taux de facteur VIII doit être augmenté au moins à 30 U/dl. Traitement des épisodes hémorragiques sévères provoqués par un
traumatisme:
Où le taux de facteur VIII doit être augmenté au moins à 50 U/dl.
Dans ces deux derniers cas, le traitement de choix, reste le concentré de facteur VIII recombinant en association avec le plasma frais congelé pour achever un taux en facteur V au dessus de 25 U/dl.
Prévention des risques hémorragiques de la chirurgie :
Le traitement consiste en une association de la DDAVP, pour maintenir le taux du facteur VIII au dessus de 50 U/dl, avec du plasma frais congelé viro-inactivé pour achever un niveau minimal du facteur V qui est de 25 U/dl. Ces deux produits doivent être administrés toutes les 12 h jusqu’à la cicatrisation.
L’association avec le concentré de facteur VIII peut être envisagée, si l’efficacité de la DDAVP semble insuffisante sur le facteur VIII34
42 Tableau 5 : Principe du traitement avec le taux des F V et VIII à atteindre
Traitement F V F VIII Périodes
hémorragiques
mineurs Plasma frais congelé + FVIII recombinant >25U/dl >30U/dl Périodes hémorragiques sévères >25U/dl >50U/dl Acte chirurgical mineur Si FVIII>5U/dl : Plasma frais congelé + DDAVP.
Si FVIII<5U/dl : Plasma frais congelé + concentré de F VIII.
>25U/dl >50U/dl
Acte chirurgical profond
≈ 70U/dl ≈ 100U/dl
2- Prise en charge de la femme enceinte et de son bébé :
a- La femme enceinte 31 :
La prise en charge des patientes atteintes du déficit combiné en FV et VIII en contexte obstétrical, nécessite la collaboration de plusieurs unités ; l’obstétricien, le centre d’hémophilie, et l’hématologue.
Le contrôle du niveau plasmatique des FV et VIII, doit se faire dans le 3ème trimestre de la grossesse.
Ce contexte obstétrical constitue un cas particulier, pour ces femmes enceintes et atteintes de ce déficit combiné, puisque le taux de FVIII
43 augmente physiologiquement pendant la grossesse, contrairement au FV qui peut augmenter ou diminuer.
Donc il existe une correction totale du taux de VIII, et par conséquent tout saignement dépendra du niveau du FV au moment et après l’accouchement, ainsi l’accouchement aura lieu sous plasma normal viro-inactivé (comme source de FV), pour maintenir un taux de FV supérieur à 15 U/dl, alors que le FVIII est maintenu au dessus de 50 U/dl durant cette période.
En cas d’un accouchement par césarienne, la section peut être exécutée, mais par prudence, l’administration du FV doit continuer jusqu’à la cicatrisation, chez les femmes ayant un taux inférieur à 15 U/dl en FV. L’anesthésie par voie péridurale, n’est permise que si les taux du FV et VIII sont, successivement supérieurs à 15 U/dl et 50 U/dl.
b- Le nouveau-né 31 :
Le diagnostic du déficit combiné en FV et VIII, chez le nouveau né, peut se faire, pendant la période néonatale, en utilisant le sang du cordon ou du sang périphérique.
Dans ce cas, les bébés affectés par ce déficit combiné en facteur V et VIII, doivent recevoir par voie orale plutôt que la voie intramusculaire de la vitamine K, et par voie sous cutanée les vaccinations de l’enfance. Les hémorragies intracrâniennes, ne sont pas décrites, dans ces conditions.
44
2
éme
chapitre :
Une nouvelle approche
de la synthèse et de la sécrétion des
facteurs V et VIII
45 I-
Maturation des facteurs V et VIII
La production de ces deux facteurs commence par la synthèse d’un polypeptide de 2224(15) aa pour le facteur V et de 2351 aa(20) pour le facteur VIII.
1- Au niveau du réticulum endoplasmique :
Les facteurs V et VIII subissent des modifications post traductionnelles (10, 16) comprenant :
Clivage du peptide signal (15, 38)
Initiation de la N-glycosylation (liaison par une β N acètylglycosamine à des résidus asparagines de la protéine) (15, 20)
Contrôle de la « qualité »; il s’agit d’un ensemble de mécanismes assurant le transport uniquement des protéines correctement pliées.
Il fait intervenir deux protéines : la calnexine et la calreticuline pour le facteur VIII. Alors que ce contrôle est assuré uniquement par la calreticuline pour le facteur V (17, 39) .
La calnexine et la calreticuline possèdent des domaines lectines ayant un rôle crucial dans leur fonction comme chaperonne (33, 39).
Une fois les facteurs V et VIII correctement pliés, assemblés et glycosylès, ils se fixent à une protéine réceptrice l’ERGIC-53/LMAN1 par l’intermédiaire de leurs résidus mannose (18)
46 spécifiques présents dans le domaine CRD (Carbohydrate Recognition Domain) de LMAN1 permettent cette liaison.
La structure cristalline de LMAN1-CRD permet d’identifié une surface de conservation des résidus dans le coté opposé du site mannose-binding qui peut servir à la fixation de MCFD2, ou d’un ligand additionnel (38), l’interaction entre LMAN1-MCFD2 avec FVIII + FV dépend de la concentration intracellulaire en calcium (33).
LMAN1 présente des séquences di aromatiques Phe-Phe (FF) qui semblent fonctionner comme un signal de sortie du réticulum endoplasmique des cofacteurs V et VIII (7, 33, 38) .
Le complexe LMAN1-MCFD2 + FV /FVIII est orienté vers des vésicules de transport de type COP II qui assure le transport antègrade (du Réticulum Endoplasmique vers l’Appareil de Golgi) de ces protéines vers le compartiment intermédiaire entre RE et Golgi (ERGIC)(7, 33, 38)
.
a- Au niveau du compartiment intermédiaire:
LMAN1 se dissocie des facteurs V et VIII, le mécanisme de cette release reste peu clair elle serait liée à:
L’action de MCFD2. La dissociation de COPII.
Diminution du pH et du calcium dans l’ERGIC (38) Mais cette dissociation est probablement liée à la diminution du pH et de la
47 concentration en calcium dans l’ERGIC, bien que le pH précis de l’ERGIC ne soit pas connu, on sait cependant que le compartiment de Golgi est plus acide que celui du Réticulum Endoplasmique (7, 38) . Le complexe LMAN1-MCFD2 libre est recyclé dans des vésicules de transport de type COP-I grâce à un motif C- terminal de type lys-lys(KK), ce qui présente un signal retrieval au Réticulum Endoplasmique.
Les facteurs V et VIII, de leur part, continuent leur chemin dans les microtubules du cis Golgi probablement grâce à COP-I.
b- Au niveau de l’Appareil de Golgi :
O-glycosylation (liaison par une α N acètylglucosamine à une serine ou à une thréonine) (20) .
Poursuite des N-glycosylations (10, 20) . Sulfatation (10, 20) .
Phosphorylation (10, 20).
Clivage endoprotéolytique du monomère de 300 KD (du facteur VIII uniquement), au niveau des résidus 1313 et 1648 pour donner un hétérodimère possédant une chaine légère (80KD), une chaine lourde (200KD) et une portion variable du domaine B. ces deux chaines sont réunies par des ponts calcium (16, 20) .
48
1- Sécrétion des facteurs V et VIII hors des cellules :
Ainsi le facteur VIII est secrété des cellules sous forme d’heterodimère, contrairement au facteur V qui est secrété sous forme d’une seule chaine d’acides aminés riches en groupements hydrophobes (15, 19)
. Une partie de 20 à 25% est stocké dans les granules alpha des mégacaryocytes (7, 15, 18)
où le FV est complexé à la multimèrine.
2- Facteurs V et VIII dans la circulation :
Une fois dans la circulation, le facteur VIII est lié au facteur de V.Willebrand par l’extrémité N- terminal de la chaine légère formant ainsi un complexe multimèrique de 1 molécule de FVIII pour 36 de facteur de Von Willebrand (F vw) (12) qui joue un rôle de protéine porteuse.
Le facteur V plasmatique n’est complexé à aucune autre protéine plasmatique (19).
49
Figure 8. Modèle de transport des FV et FVIII entre le réticulum endoplasmique
(RE) et l'appareil de Golgi, selon Zhang et Ginsburg [36].Les facteurs V et VIII se fixent au complexe LMAN1–MCFD2 par l'intermédiaire de leurs résidus mannose. Ce complexe LMAN1–MCFD2 + FV/FVIII est orienté vers les vésicules de transport par les protéines COPII. Ces vésicules arrivent au niveau de l'appareil de Golgi et relarge le facteur
de la coagulation (FV ou FVIII) dans l'appareil de Golgi, à partir duquel son métabolisme intracellulaire se poursuivra jusqu'au phénomène de sécrétion. Le complexe LMAN1– MCFD2 est lui recyclé grâce à la prise en charge par les protéines COP I qui dirigent le complexe transporteur dans des vésicules de transport antérogrades, d'où son retour vers le
50
2éme partie :
51 I- Antécédents et histoire familiale :
FZ est une enfant de 13 ans, de sexe féminin, de parents consanguins, sans antécédents cliniques adressée au laboratoire d’hématologie pour un bilan préopératoire.
II- Expression clinique :
La jeune fille présentait une verrue sur la lèvre qu’elle devait faire enlever dans un cabinet privé d’ORL qui nous l’a adressée.
III- Diagnostic biologique : Le bilan biologique a montré :
Un hémogramme normal.
Le bilan d’hémostase révèle un allongement du TCA 81’’/35’’ combiné à celui du TP 34%.
Le TS, le fibrinogène et le taux PTL sont normaux.
Le dosage différentiel des facteurs du temps de quick et du TCA ont permis de révéler un déficit combiné en FV (9%), et en FVIII (9%). Les facteurs VII, X, IX, XI, XII et II sont sans particularité.
Le diagnostic d’un déficit combiné en facteur V et VIII est retenu.
Le bilan d’hémostase des deux parents n’a révélé aucune anomalie : le dosage des FV et FVIII sont normaux
52
Mère Père
FV 82% 104%
FVIII 94% 116%
Tableau 6 : Enquête familiale
Le déficit combiné en FV/FVIII est en effet une maladie à transmission autosomique récessive.
La famille a malheureusement refusé de nouveaux prélèvements et leur envoi à l’étranger pour une enquête moléculaire.
53
54 Le déficit combiné en FV et VIII est une pathologie rare, mais vu sa répartition Méditerranéenne, il faut y penser surtout devant des enfants présentant des saignements mineurs avec un TQ et un TCA allongés.
Néanmoins cette maladie reste très peu diagnostiquée, soit par l’absence de signes cliniques graves induisant une absence de consultation médicale, soit par un diagnostic d’hémophilie A mineure dont le TQ est légèrement allongé par excès chez les sujets masculins atteints de ce déficit. Pour cette raison un dosage du FV doit être réalisé pour tout déficit modéré en FVIII.
L’étude de ce déficit combiné a permis la mise en évidence d’une voie de transport intracellulaire des FV et VIII. Pour autant on peut se demander si ce système est exclusivement réservé à ces deux facteurs de coagulation, ou y a t’il d’autres protéines qui ont une sécrétion par cette voie ? Il est fort probable qu’il y en a, mais dont l’altération n’a aucune répercussion clinique ou biologique.
Les concentrations des autres protéines plasmatiques étant normales, alors que celles des FV et VIII étant variables mais non nulles, suggérant que ce mode de transport de ces deux facteurs reste l’exemple le plus connu mais probablement pas le seul, et il est fort probable qu’un transport résiduel de ces deux facteurs existe indépendamment du complexe LMAN1-MCFD2.
En outre, les détails du mécanisme de l’identification, du transport et de la libération des cargos (protéines) par LMAN1 et MCFD2 sont mal compris (7). La sortie et le transport continus des protéines de l’ERGIC vers l’Appareil de Golgi sont aussi mal compris, et il sera intéressant de savoir s’il existe des
55 récepteurs spécifiques des protéines cargo (FV et VIII) pour le transport antègrade de l’ERGIC vers l’appareil de Golgi(7)
.
De plus on note toujours la méconnaissance actuelle de la cause du déficit combiné chez certaines familles souffrant de ce déficit combiné sans aucune anomalie dans les gènes de ces deux protéines (LMAN1 et MCFD2).
Ce mécanisme de sécrétion des facteurs V et VIII de la coagulation reste donc incomplet. La poursuite de l’étude de ces familles permettra dans les années qui viennent d’élucider la totalité de ce phénomène.
Néomoins ces connaissances ont permis d’ouvrir la voie vers la recherche de médicaments anti thrombotiques dans les hyper sécrétions du FVIII prédisposant à des accidents thromboemboliques ou encore dans le FV Leiden.
Parmi les nombreux facteurs prédisposant à la thrombose veineuse, deux concernent les facteurs V et VIII.
Augmentation du taux circulant de facteur VIII.
Mutation Leiden du gène du facteur V induisant la production d’un facteur V résistant à l’inactivation de la protéine C.
Un blocage partiel de la synthèse des facteurs V et VIII par inhibition de LMAN1 et/ou de MCFD2 pourrait être une cible innovante d’une thérapeutique anticoagulante, particulièrement chez les sujets présentant ces facteurs biologiques de risque de maladie thromboembolique veineuse (36).
56
Résumé
Le déficit combiné en facteur V et VIII est décrit depuis 1954 comme une maladie congénitale rare. Il se répartit principalement dans le contour méditerranéen et se distingue des déficits classiques des facteurs de la coagulation.
La description des familles atteintes de ce déficit combiné a permis dans un premier temps de connaître la symptomatologie de la maladie, puis d’identifier les gènes responsables de la diminution simultanée des FV et VIII.
Deux gènes codent successivement pour deux protéines LMAN1pour Lectin MANnose-Binding1 et MCFD2 pour Multiple Coagulation factor deficiency2 qui sont impliquées dans le passage des FV et VIII entre le réticulum endoplasmique et l’appareil de Golgi avant la sécrétion.
Les mutations décrites dans le gène de LMAN1 (situé sur le chromosome 18q21) et dans le gène de MCFD2 (situé sur le chromosome 2p21) entravent la structure des molécules chaperonnes et provoquent un déficit combiné en facteur V et VIII.
Le mécanisme une fois élucidé a permis d’ouvrir des voies de recherche pour la prise en charge des thromboses liées à l’augmentation du FVIII. Nous rapportant le premier cas marocain de ce déficit, il s’agit d’une jeune petite fille de 13 ans, originaire de Khemissat, sans antécédents cliniques, adressée au laboratoire pour un bilan préopératoire. L’hémogramme est normal, le bilan d’hémostase a révélé une diminution du TP (34%) et un
57 allongement du TCA (81s/ 35s) liés à un déficit combiné en FV et VIII dont les taux successifs se sont révélés identiques et égaux à 9%. le reste de l’hémogramme est normal.
58
59 1- Guillin M. C.
- Déficits constitutionnels des facteurs Il, V, VII ou X.
-Encycl.Med.Chir. (Paris- France), Sang, 13021ClO, 7-1988,6p
2- Dipika Mohanty, Kanjaksha Ghosh, Shrimati Shetty, Marta Spreafico, Isabella Garagiola, and Flora Peyvandi
Mutations in the MCFD2 Gene and a Novel Mutationin the LMAN1 Gene in Indian Families with Combined Deficiency of Factor V and VIII American Journal of Hematology 79:262-266 (2005)
3- Beat Nyfeler, Yukiko Kamiya, Françoise Boehlen , Kazuo Yamamoto4, Koichi Kato Philippe de Moerloose, Hans-Peter Hauri and Marguerite Neerman-Arbez
Deletion of three residues from the C-terminus of MCFD2 affects binding to ERGIC-53 and causes combined factor V and factor VIII deficiency.
Blood First Edition Paper, prepublished online October 30, 2007
4- M. Neerman-Arbez, K.M. Johnson, M.A. Morris, J.H. McVey, F. Peyvandi, W.C.
Molecular Analysis of the ERGIC-53 Gene in 35 Families With Combined Factor V-Factor VIII Deficiency
Blood, Vol. 93 No. 7, 1999
5- Bin Zhang, Beth McGee, Jennifer S. Yamaoka, Hugo Guglielmone, Katharine A. Downes, Salvador Minoldo, Gustavo Jarchum,Flora
60 Peyvandi, Norma B. de Bosch, Arlette Ruiz-Saez, Bernard Chatelain, Marian Olpinski, Paula Bockenstedt, Wolfgang Sperl, Randal J. Kaufman, William C. Nichols, Edward G. D.Tuddenham, and David Ginsburg
Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2
BLOOD, 2006 vol 107, N°5
6- Ginsburg D, Nichols WC, Zivelin A, Kaufman RJ, Seligsohn U Combined factors V and VIII deficiency-the solution.
Haemophilia. 1998 (4).
7- Andrea C. Baines and Bin Zhang
Receptor-mediated protein transport in the early secretory pathway Biochemical Sciences 2007 Vol.32 No.8
8- Kimberly A. Marquette, Debra D. Pittman, and Randal J. Kaufman.
The Factor V B-Domain Provides Two Functions to Facilitate Thrombin Cleavage and Release of the Light Chain.
Blood, Vol 86, N° 8. 1995.
9- Timothy Myles, Thomas H. Yun, and Lawrence L. K. Leung.
Structural requirements for the activation of human factor VIII by thrombin.
Blood, 2002 vol 100, N° 8.
10- Kenneth G. Mann and Michael Kalafatis
Factor V: a combination of Dr Jekyll and Mr Hyde. Blood, 2003 VOL 101
61 11- DENNIN GERMH ET HUISSE MG.
Affections hémorragiques par anomalie congénitale ou acquise de la coagulation (en dehors de l'hémophilie et de la maladie de willebrand). Encycl. Med Chir (Elsevier-Paris), Hématologie, 13-021-C-10, 1997,12p 12- GIRODONE, GAZENGEL C ET GOOSSENS M.
Aspects moléculaires des hèmophilies.Encyci Med Chir (Paris-France), Hématologie, F.a.13-021-B-10, 1995,8p.
13- P. Vincent Jenkins, Jan Freas, Kyla M. Schmidt, Qian Zhou, and Philip J. Fay.
Mutations associated with hemophiliaAin the 558-565 loop of the factor VIlla A2 subunit alter the catalytic activity of the factor Xase complex. Blood, 2002 _ VOL 100, N° 2.
14- Michèle Gouault-Heilmann. Physiologie de la coagulation.
Aide-mémoire d'hémostase.2ème édition 2000.
15- Kenneth Segers, Bjorn Dahlback, Gerry A. F. Nicolaes
Coagulation factor V and thrombophilia: Background and mechanisms. Journal of Thrombosis and Haemostasis 2007; 98
16- Richard van Wijk, Karel Nieuwenhuis, Marijke van den Berg, Eric G. Huizinga, Brenda B. van der Meijden, Rob J. Kraaijenhagen and Wouter W. van Solinge.
Five novel mutations in the gene for human blood coagulation factor V associated with type 1 factor V deficiency.
Blood, 2001 VOL 98, N°2.
62 Differentiel Interaction of Coagulation Factor VIII and V with Protein Chaperone Calnexin and Calreticulin.
The journal of biological chemistry vol.273, N°14, 1998.
18- Micheline Moussalli, Steven W. Pipe,Hans-Peter Haurii, William C. Nichols,David Ginsburg and Randal J. Kaufman
Mannose-dependentEndoplasmic Reticulum(ER)-Golgi Intermediate
Compartment-53-mediatedER to Golgi Trafficking ofCoagulation Factors V and VIII
The journal of biological chemistry Vol. 274, N° 46, 1999.
19- Debra D. Pittman, Kimberly A. Marquette, and Randal J. Kaufman Role of the B Domain for Factor VI11 and Factor V Expression
and Function
Blood, Vol 84, N° 12. 1994.
20- Peter J. Lenting, Jan A. van Mourik, and Koen Mertens
The Life Cycle of Coagulation Factor VIII in View of Its Structure and Function.
Blood .1998, Vol 92, N° 11
21- Ty E. Adams, Matthew F. Hockin, Kenneth G. Mann, and Stephen J. Everse.
The crystal structure of activated protein C-inactivated bovine factor Va. PNAS, 2004 vol 101 N°24.
22- Hongzhi Z. Miao, Nongnuch Sirachainan, Lisa Palmer, Phillip Kucab, Michael A. Cunningham, Randal J. Kaufman, and Steven W. Pipe
63 Bioengineering of coagulation factor VIII for improved secretion
Blood, 2004 vol 103, N° 9
23- Eva Ajzner, Istvan Balogh, Terez Szabo, Aniko Marosi, Gizella Haramura, and Laszlo Muszbek.
Severe coagulation factor V deficiency caused by 2 novel frameshift mutations.
Blood, 2002 vol 99, N° 2.
24- OMIM=www .ncbi .nlm.nih.gov/entrez/quary.fcgi ?db Factor V and Factor VIII, combined deficiency of ; F8F5D 25- U. SELIGSOHN and D. GINSBURG
Deciphering the mystery of combined factor V and factor VIII deficiency
Journal of Thrombosis and Haemostasis, 4, 2006. 26- Pier Mannucci, Stefano Duga, and Flora Peyvandi
Recessively inherited coagulation disorders Blood, 2004Vol.104, N° 5
27- William C. Nichols, Uri Seligsohn, Ariella Zivelin, Valeri H. Terry, Colette E. Hertel, Matthew A. Wheatley, Micheline J. Moussalli, Hans-Peter Hauri, Nicola Ciavarella, Randal J. Kaufman, and David Ginsburg.
Mutations in the ER–Golgi Intermediate Compartment Protein ERGIC-53 Cause Combined Deficiency of Coagulation Factors V and VIII Cell, Vol. 93, 1998.
28- William C. Nichols, Uri Seligshon, Ariella Zivellin, Valeri H, Terry, Nathan D. Arnold, David R. Siemieniak, Randal J. Kaufman, and
64 David Ginsbueg
Linkage of Combined Factor V and VIII Deficiency to Chromosome 18q by Homozygosity Mapping
Journal of Clinical Investigation. vol99, n°4, 1997.
29- N.Sirachainan, B.Zhang, A.Chuansumrit, S.Pire, W.Saankuls and D.Ginsburg
Combined factor V and factor VIII deficiency in a Thai patiente: a case report of genotype and phanotype.
Haemophilia 2005
30- www.med.unc.edu/isth/mutationsdatabases/FVandVIII_2007.htm 2007mutation in patients with combined factor V and VIII deficiency 31- P.H.B.Bolton-Maggs, D.J.Perry, E.A.Chalmers, L.A.Parapia, J.T
Wilde, M.D.Williams, P, W, Collins, S.Kitchen, G.Dollan and A.D.Mumford
The rare coagulation disorder- review with guidelines for management from the United Kingdom Haemophilia Centre Doctor’s Organisation. Heamophilia, 10, 2004
32- William C. Nichols, Valeri H. Terry, Matthew A. Wheatley, Angela Yang, Ariella Zivelin, Nicola Ciavarella,Caterina Stefanile, Tadashi Matsushita, Hidehiko Saito, Norma B. de Bosch, Arlette Ruiz-Saez,Argimiro Torres, Arthur R. Thompson, Donald 1. Feinstein, Gilbert C. White, Claude Negrier, Christine Vinciguerra, Melih Aktan, Randal J. Kaufman, David Ginsburg, and Uri Seligsohn ERGIC-53 Gene Structure and Mutation Analysis in 19 Combined Factors V and VIII Deficiency Families
65 Blood, vol 93, (1999).
33- Hans-Peter Hauri, Felix Kappeler, Helena Andersson and Christian Appenzeller
ERGIC-53 and traffic in the secretory pathway Journal of Cell Science 113, 587-596 (2000)
34- Hans-Peter Hauri, Christian Appenzeller, Fransiska Kuhn, Oliver Nufer.
Lectins and trafic in the secretory pathway FEBS Letters 476 (2000)
35- Bin Zhang, Michael A Cunningham, William C Nichols, John A Bemat, Uri Seligsohn, Steven W Pipe, John H McVey, Ursula Schulte-Overberg, Norma B de Bosch, Arlette Ruiz-Saez, Gilbert C. White, EGD Tuddenham, Randal J. Kaufman & David Ginsburg Bleeding due to disruption of a cargo-specifie ER-to-Golgi transport complex
NATURE GENETICS 2003 Vol 341 N°2 36- C. Vinciguerra, B. Durand and L. Rugeri
Déficit combiné en facteurs V et VIII de la coagulation : ou quand la génétique nous explique les déficits combinés de facteurs de la coagulation
Immuno-analyse & Biologie Spécialisée Vol 22, 2007
37- Elena M. Faioni, Gessica Fontana, Giovanni Carpani, Enza D'Auria, Giuseppe Banderali,Gianalessandro Moronib, Marco Cattaneo Review of clinical, biochemical and genetic aspects of combined factor V and factor VIII deficiency, and report of a new affected family
66 Thrombosis Research 112 (2003)
38- Zhang B, Ginsburg D.
Familial multiple coagulation factor deficiencies :new biologic insight from rare genetic bleeding disorders.
Journal of Thrombosis and Haemostasis 2004
39- Joseph D. Schrag, Daniela O. Procopio, Miroslaw Cygler, David Y.Thomas and John J.M.Bergeron.
Lectin control of protein folding and sorting in the secretory pathway. Biochemical Science 2003 Vol.28 N° 1
40- Roula A. Farah, philippe de MOerloose, Isabelle Bouchardy, Michael A, Morris, wadad Barakat, Alain E. Sayad, Marguerite Neerman-Arbez
Combined factor V – factor VIII deficiency (F8F5D): Compound heterozygosity for two novel truncating mutations in LMAN1in a consanguineous pateint
Journal of Thrombosis and Haemostasis 2006; 95
41- Chantal Arar, Valèrie Carpentier, Jean- Pierre Le Caerl, Michel
Monsigny, Alain Legrand, and Annie- Claude Roche.
ERGIC- 53, a menbrane protein of the Endoplasmic Reticulum-Golgi Intermediate Compartiment, Is Identical to MR60, an Intracellular Mannose-specific Lectin of Myelomonocytic Cells.
Biochemistry and Molecular Biology ;Vol 270; N°8, 1995
42- Oliver Nufer, Felix Kappeler, Svend Gulbrandsen and hans- Peter Hauri
67
ER export of ERGIC-53 is controlled by cooperation of targeting determinants in all three of its domains.
Journal of Cell Science; 116; 2003.
43- Miyoko Higuchi, Haig H. Kazazian, Jr, Laura Kasch, Tina C. Warren, Matthew J. McGinniss, John A. Phillips III, Carol Kasper, Robert JanKo, and Stylianos E. Antonarakis.
Molecular characterization of svere hemophilia A suggests that about half the mutations are not within the coding regions and splice junction of the factor VIII gene.
Genetic; vol 88; 1991.
44- Etienne P.A.Neve, Kestin Svensson, Jonas Fuxe, and Ralf F. Pettersson.
VIPL, a VIP36-like membrane protein with a putative function In the
export of glycoproteins from the endoplasmic reticulum Experimental Cell Research 288 (2003)
45- William C. Nichols and David Ginsburg
PROTEIN BIOSYNTHESIS’99 From the ER to the Golgi: Insights from the study of combined Factor V and VIII Deficiency
American journal of human genetic; 46; 1999
46- Kevin R. Viel, Deepa K. Machiah, Diane M. Warren, Manana Khachidze, Alfonso Buil, Karl Fernstrom, Juan C.Suoto
A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and assoaciations with plasma FVIII activity levels






![Figure 8. Modèle de transport des FV et FVIII entre le réticulum endoplasmique (RE) et l'appareil de Golgi, selon Zhang et Ginsburg [36]](https://thumb-eu.123doks.com/thumbv2/123doknet/14384095.700136/49.892.228.671.239.527/figure-modèle-transport-fviii-réticulum-endoplasmique-appareil-ginsburg.webp)

