HAL Id: hal-03065695
https://hal.archives-ouvertes.fr/hal-03065695
Submitted on 16 Dec 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of
sci-entific research documents, whether they are
pub-lished or not. The documents may come from
L’archive ouverte pluridisciplinaire HAL, est
destinée au dépôt et à la diffusion de documents
scientifiques de niveau recherche, publiés ou non,
émanant des établissements d’enseignement et de
Synthesis of BEC-type germanosilicates with
asymmetric diquaternary ammonium salts
Carole Isaac, Jean-Louis Paillaud, T. Jean Daou, Andrey Ryzhikov
To cite this version:
Carole Isaac, Jean-Louis Paillaud, T. Jean Daou, Andrey Ryzhikov. Synthesis of BEC-type
ger-manosilicates with asymmetric diquaternary ammonium salts. Microporous and Mesoporous
Materi-als, Elsevier, 2020, 312, pp.110804. �10.1016/j.micromeso.2020.110804�. �hal-03065695�
Synthesis of BEC-type germanosilicates with
asymmetric diquaternary ammonium salts
Carole Isaac, Jean-Louis Paillaud∗, T. Jean Daou, Andrey Ryzhikov
Universit´e de Haute-Alsace (UHA), CNRS, Institut de Science des Mat´eriaux de Mulhouse (IS2M), UMR 7361, 68100 Mulhouse, France.
Universit´e de Strasbourg, 67000 Strasbourg, France.
Abstract
This paper deals with the synthesis of germanosilicate zeolites using asymmet-ric diquaternary ammonium salts as organic structure-directing agents (OSDA). Seven pyrrolidinium and piperidinium derivatives were chosen, with different carbon chain lengths and different terminal groups linked to the nitrogen atoms. For each OSDA, the syntheses were performed with four different gel compo-sitions, i.e. in fluoride or hydroxide media both in concentrated or diluted conditions. The products were identified using powder X-ray diffraction data (PXRD). The integrity of the structure-directing agents were verified using13C
liquid and solid-state NMR spectroscopy and thermogravimetric analyses. For one sample, the Rietveld method allowed us to localize the OSDA inside the pore and the fluoride ions inside the d4r composite building unit but, also inside the main 12-ring channels as proved by19F solid-state NMR spectroscopy.
Keywords: Zeolite framework-type BEC, hydrothermal synthesis, organic structure-directing agents, Rietveld refinement,19F MAS NMR
∗Corresponding author
1. Introduction
There has been a growing interest for the synthetic zeolites since their first exploitation in industry [1]. Research on the synthesis of zeolites has intensi-fied since the 1940s, with the discovery of new zeolitic phases without natural equivalents (zeolites A [2]). Syntheses were done at autogeneous pressure and 5
high temperature in alkaline medium to reproduce their natural conditions of crystallization. It was only from the 1960s onwards, following the founding work of Barrer [3], that using organic structure-directing agents (OSDAs) have become widespread. Organic templates like quaternary ammonium salts have speeded up research and led to enhance the silica content [4, 5,6]. This major 10
advance allowed the discovery of many new topologies such as β zeolite [7] as well as the possibility of incorporating more silicon as T atoms, thus allowing the synthesis of the first all-silica zeolite in 1977 [8]. Among the vast family of ammoniums used as OSDA, the symmetric diquaternary alkylammonium salts of general formula R3N+(CH
2)nN
+R
3 have been widely reported, leading to
15
a variety of zeolites, depending in particular on the length of the polymethy-lene chain and the nature of the R alkyl group. In Tables 1 and 2 are listed some zeolites obtained in pure silica, (Si,Al) and (Si,Ge) media using this type of OSDAs. From these data, it is rather difficult to learn about the true role of the structuring OSDAs. However, it appears that for polymethylene chains 20
with less than five carbons and with similar terminal groups in terms of steric hindrance in (Si,Al) media, small and medium pore zeolites were discovered. The short imidazolium based dication, 3,3’-(propane-1,3-diyl)bis(1,2-dimethyl-1H -imidazol-3-ium) with a shorter propyl linking chain leads to HPM-12 [9], a disordered silicogermanate of topology *UOE [10]. The addition of a car-25
bon in the alkyl chain, (3,3’-(butane-1,4-diyl)bis(1,2-dimethyl-1H -imidazol-3-ium) leads to STW zeolites in pure silica and (Si,Ge) media [11]. In (Si,Al) synthetic media with an excess of fluoride, similar bis-pyrazolium derivatives PST-22 (PWW) and PST-30 (PTY) were produced [12] (see Table 1). Sub-stitution of methyl by ethyl groups, the symmetric OSDAs of chemical formula 30
(H5C2)3N+(CH 2)nN
+(C
2H5)3 where n=3-10 (not reported in Table 2 except
for the N1,N1,N1,N4,N4,N4-hexaethylbutane-1,4-bis(aminium) derivative)
com-bined with different sources of alkaline cations (Li+, Na+or K+) induce the
crys-tallization of P1 (GIS), ZSM-5 (MFI), mordenite (MOR), ZSM-50 (EUO), ZSM-57 (MFS), SUZ-4 (SZR) [13] and SSZ-16 (AFX) [14] zeolites.
35
A second major discovery, initiated by Flanigen, [15] then used by Guth et al. [16], was made at the end of the 70s with the introduction of the fluoride anions as mineralizing agents (F−) instead of OH−. By using the fluoride ion as a mineralizer (the so-called ”fluoride route”), it is possible to perform the synthesis in solutions which are less supersaturated in silica-based species and 40
the pH of which is between 5 and 9 [17]. This method has enabled the discovery of many new pure silica microporous materials such as e.g. octadecasil [18] and ITQ-3 [19], two early examples.
The third major advance in zeolite synthesis was the introduction of hetero-elements such as iron, gallium, titanium and germanium. In germanium-con-45
taining zeolitic framework, i.e. with tetrahedrally coordinated T atoms, the angle between Ge-O-Ge bonds is smaller than the one of Si-O-Si bonds so the formation of smaller units such as double 4-ring (d4r ) is enhanced and allows a greater structural diversity [20, 21, 22]. The use of N1,N1,N1,N6,N6,N6 -hexamethylhexane-1,6-bis(aminium) as OSDA leads to IM-10 [23] (UOZ), a 50
germanate or silicogermanate with a clathrasil-like structure. Four additional carbons in the central alkyl chain were needed to obtain IM-17 [24] (UOV), a germanosilicate with a 3D pore system with 12-, 10- and 8-ring pore-openings (see Table 2). Here, the alkyl chain length makes the difference between a clathrasil and an open framework structure.
55
In this article, we report about the synthesis of zeolites using asymmetric di-quaternary ammonium salts as OSDAs. Seven pyrrolidinium and piperidinium derivatives were chosen, with different carbon chain lengths and different ter-minal groups linked to the second nitrogen atom (see Table3). Our intention was to verify if the asymmetry directs or not to the syntheses of new topologies 60
and to see an eventual influence of the asymmetry on the obtained structures. For each OSDA, the syntheses were performed in the (Si,Ge) system with four different gel compositions (fluoride or hydroxide media both in concentrated or diluted conditions).
Table 1: Some symmetric diazolium-based imidazolium [I.] and (pyrazolium [P.] deriva-tives) salts used as OSDAs in zeolite synthesis.
(I., P.)N+(CH 2)nN
+(I., P.) Obtained phases
Si (Si,Al) (Si,Ge) Ref.
N + N + N N 1 (*UOE)HPM-12 [9, 10] N + N + N N 2 STW STW [11] N N+ + N N 3 TON [25] NN+ + N N 4 PST-22 (PWW)8 [12] NN+ + N N 5 PST-30 (PTY)8 [12] NN+ + N N 6 PST-22 (PWW)8 [12] N N+ + N N 7 PST-22 (PWW)8 [12]
13,3’-(propane-1,3-diyl)bis(1,2-dimethyl-1H -imidazol-3-ium),2 3,3’-(butane-1,4-diyl)bis(1,2-dimethyl-1H -imidazol-3-ium), STW also with the -(CH2)5- and -(CH2)6- derivatives, 3 3,3’-(butane-1,4-diyl)bis(1-methyl-1H -imidazol-3-ium), TON also with the -(CH2)5- and -(CH2)6- derivatives, 4 2,2’-(butane-1,4-diyl)bis(1-methyl-1H -pyrazol-2-ium) also with the -(CH2)5- derivative, 5 2,2’-(butane-1,4-diyl)bis(1,3-dimethyl-1H -pyrazol-2-ium), 6 2,2’-(butane-1,4-diyl)bis(1,4-dimethyl-1H -pyrazol-2-ium), 7 2,2’-(butane-1,4-diyl)bis(1,5-dimethyl-1H -pyrazol-2-ium),8 Synthesis performed under the excess fluoride conditions.
Table 2: Some zeolites produced with symmetric diquaternary alkylammonium salts as OSDAs.
R3N+(CH2)nN+R3
Obtained phases1
Si (Si,Al) (Si,Ge) Ref.16
N+ (CH 2)5 N+ 2 ZSM-48 (*MRE) ZSM-50 (EUO) [26,27] N+ (CH 2)6 N+ 3 ITQ-13, IM-7 (ITH) ZSM-50 Eu-1 (EUO) ITQ-17 (BEC) IM-10 (UOZ) [23,28,29,30] N+ (CH 2)9 N+ 4 β (*BEA), Silicalite-1 (MFI) ZSM-12 (MTW) [26,27] N+ (CH2)10N+ 5 β (*BEA) Nu-87 NES IM-17 (UOV) [24,26,31] N+ (CH 2)4 N+ 6 TNU-9 (TUN) TNU-10 (STI) [32,33] N+ (CH 2)5 N+ 7 IM-5 (IMF) [34,35] N+ (CH 2)6 N+ 8 SSZ-74 (-SVR) EMM-26 (EWS)14 [36,37] N+ (CH 2)5 N+ 9 IZM-3 [38] N+ (CH 2)6 N+ 10 IZM-2 IZM-2 [39,40] N+ (CH 2)4 N+ 11 SSZ-16 (AFX) [14] N+ (CH 2)4 N+ N N 12 SSZ-16 (AFX) BEC [14,41] N+ (CH 2)4 N+ 13 ITQ-14 (β-BEC overgrowth) SSZ-16 (AFX)15 BEC [14,41,42]
1 In fluoride and/or OH− media, 2 N1,N1,N1,N5,N5,N5 -hexamethylpentane-1,5-bis(aminium), 3 N1,N1,N1,N6,N6,N6-hexamethylhexane-1,6-bis(aminium), 4 N1,N1,N1,N9,N9,N9-hexamethylnonane-1,9-bis(aminium), 5 N1,N1,N1,N10,N10,N10 -hexamethyldecane-1,10-bis(aminium), 6 1,1’-(butane-1,4-diyl)bis(1-methylpyrrolidin-1-ium), 7 1,1’-(pentane-1,5-diyl)bis(1-methylpyrrolidin-1-ium), 8 1,1’-(hexane-1,6-diyl)bis(1-methylpyrrolidin-1-ium), 9 1,1’-(pentane-1,5-diyl)bis(1-methyl-piperidin-1-ium), 10 1,1’-(hexane-1,6-diyl)bis(1-methyl-piperidin-1-ium),11 N1,N1,N1,N4,N4,N4 -hexaethylbutane-1,4-bis(aminium),12 1,1’-(butane-1,4-diyl)bis(1,4-azabicyclo[2.2.2]octan-1-ium),13 1,1’-(butane-1,4-diyl)bis(1-azabicyclo[2.2.2]octan-1-ium), 14 EMM-26 is a borosilicate zeolite, 15 also synthesized with 8,8’-(butane-1,4-diyl)bis(8-methyl-azabicyclo[3.2.1]octan-8-ium), 16 The derivatives N1,N1,N1,Nn,Nn,Nn-n-ane-1,n-bis(aminium) with n = 7, 8, 11, 12 lead to zeolite ZSM-23 (Si,Al) MTT and for n = 14 to zeolite ZSM-12 (Si,Al) MTW [27].
2. Experimental 65
2.1. Synthesis
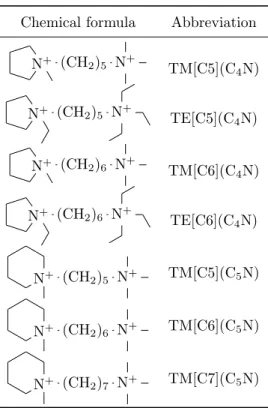
The reactants used for the zeolite syntheses were tetraethoxysilane (TEOS, Aldrich, 98 %), germanium dioxide (Aldrich, 99.99 %), hydrofluoric acid (Norma-pur, 40 %) and the diquaternary ammonium iodine salts (OTAVA Chemicals) presented in Table3. The pyrrolidinium-derivatives (C4N) have a triethylamine
70
(TE) or trimethylamine group (TM) and a linear chain composed of five or six carbons ([C5], [C6]). The piperidinium derivatives (C5N) have a carbon chain
from five to seven atoms length.
The commercial diquaternary ammonium salts were exchanged as hydroxides using an excess of DowexTMMonosphereTM 550 A UPW (OH) resin (Supelco).
75
The concentration of the solutions was determined by 1H liquid NMR
spec-troscopy using a 0.5 mol L−1 solution of dioxane in deuterium oxide as internal standard. The conversion rate was determined by titrating the hydroxide ions with an aqueous solution of hydrochloric acid 10−2mol L−1.
Table 3: List of the structure-directing agents used in this work.
Chemical formula Abbreviation N+ (CH2)5 N+ TM[C5](C4N) N+ (CH2)5 N+ TE[C5](C 4N) N+ (CH2)6 N+ TM[C6](C4N) N+ (CH2)6 N+ TE[C6](C 4N) N+ (CH 2)5 N+ TM[C5](C5N) N+ (CH 2)6 N+ TM[C6](C5N) N+ (CH 2)7 N+ TM[C7](C5N)
For each structure-directing agent of global formula (R(OH)2), four
compo-80
sitions of the reaction gel solution of dioxane in deuterium oxide as an internal were tested with the molar proportions presented in Table4. For the hydrother-mal syntheses, the reactants were directly introduced in the 2.5 mL Teflon R
liner. The germanium dioxide was dissolved in the OSDA solutions and 2 mmol of TEOS is added under stirring. The ethanol formed by the hydrolysis of 85
TEOS and the potential excess of water were evaporated for one day at room temperature. Some water and hydrofluoric acid can be added to reach the de-sired gel ratios. The liners containing the reaction mixture were introduced into a stainless-steel multi-autoclave, the hydrothermal treatment being performed at 170◦C for one week. The solids were recovered by filtration, washed several 90
times with distilled water and ethanol and finally dried overnight in an oven at 60◦C.
Table 4: Molar composition of the starting synthesis gels tested with each template (R(OH)2) presented in Table3.
Name SiO2 GeO2 R(OH)2 HF H2O
Conc.HF 0.8 0.4 0.3 0.6 5.0
Dil.HF 0.8 0.4 0.3 0.6 30
Conc.OH 0.8 0.4 0.3 / 5.0
Dil.OH 0.8 0.4 0.3 / 30
2.2. Characterization
Synthesized products were characterized by powder X-ray diffraction, the patterns were recorded on a STOE STADI-P diffractometer equipped with 95
a linear position-sensitive detector and a curved germanium (111) primary monochromator (CuKα1 radiation). Measurements were achieved for 2θ
an-gle values in the 5–35◦range, step 0.01◦(2θ), (PSD step = 0.1◦, time/step = 65 s). For the structural study, sample 9 was used due to its high purity (see section3.2of the discussion). Two scans were collected at room temperature in 100
the 4–89.99◦range, step 0.01◦, (PSD step = 0.1◦, time/step = 120 s), the data collection duration amounted to 55 hours and 23 minutes. For better statistics on the collected data, both scans were averaged.
The morphology and size of the crystals were determined by scanning elec-tron microscopy (SEM) with a Philips XL 30 FEG microscope equipped with 105
a microprobe Si(Li) Oxford Inca Energy analyser used to determine the Si/Ge molar ratios. Thermogravimetric analyses were performed with a METTLER Toledo thermogravimetric analyzer between 30◦C and 800◦C, the temperature was increased at a constant rate of 5◦C/ min up to 800◦C.
13C and19F MAS NMR experiments were performed on a Bruker DSX 400
110
spectrometer. The recording conditions are given in Table5. Then liquid spec-tra of the OSDAs were recorded on a Bruker Avance Neo 500 spectrometer in deuterium oxide. 13C spectra were made at 125.8 MHz with 1682 scans (having an acquisition time of 1.0879 s) and1H spectra at 500.1 MHz with 16 scans (with an acquisition time of 3.2768 s).
Table 5: Recording conditions of the MAS NMR spectra.
19F 13C
Chemical shift standard CFCl3 TMS
Frequency (MHz) 376.49 100.63 Pulse width (µs) 4 5.9 Flip angle π/2 π/2 Recycle time (s) 30 4 Spinning rate (kHz) 12 12 Number of scans 360 8000
3. Results and discussion 3.1. Synthesis
The obtained germanosilicates are listed in Table6, the corresponding XRD patterns are shown in Figures S4 to S10. In general, the structure-directing role of OSDAs is difficult to appreciate, however, some trends stand out with regard 120
to the organic molecule size and the synthesis medium.
The products synthesized with the OSDAs having the shortest carbon chain length ([C5]) produced some clathrasil-type structures like AST and UOZ. However, usual synthesis of silicogermanate or germanateAST includes tetram-ethylammonium, quinuclidine or DABCO as templates [26,43] which are much 125
smaller in size than TM[C5](C4N) or TM[C5](C5N). It can be supposed that
under such hydrothermal conditions, the corresponding molecules are decom-posed, allowing the formation of the AST phase. Also, it is known that IM-10 (UOZ) [23] can be synthesized only from a germanium-rich gel in fluo-ride media with hexamethonium (N1,N1,N1,N6,N6,N6
-hexamethylhexane-1,6-130
bis(aminium) in Table 2), an OSDA close to TM[C5](C4N) in terms of steric
hindrance.
The influence of the medium on crystallization (dilution and nature of the mineralizing agents), underlined in Figure S1 can also be observed. IM-17 (UOV) is formed only in hydroxide media in accordance with the literature 135
[24]. In diluted media, three of the tested OSDAs lead to germanium-containing silicalite-1 (MFI). In contrast, zeolite β has only crystallized in concentrated media. The formation of BEC phase is observed for all the OSDAs, but in various medium, mostly in the presence of fluoride ions.
Many of these products are multi-phasic, e.g. *BEA, polymorphs of zeolite 140
β, and polymorph C (BEC) have crystallized together in samples 5, 7, 15 and 27. Zeolite β was originally prepared using tetraethylammonium hydroxide as OSDA in the (Si,Al) system [7] and since 1967, a lot of other OSDAs have enabled its synthesis. Those given in Table2are no exception to this trend. It is worth noting that this zeolite used in several catalytic reactions may be also 145
prepared in a OSDA-free medium, using a seeding process [44]. The structure of zeolite β is complex, it is an intergrowth of polymorphs A, B and C [45].
Pure polymorph C (topology BEC) was first synthesized as pure germanate (FOS-5) in 2000 with a mixture of pyridine and 1,4-diazabicylclo-[2,2,2]octane (DABCO) as OSDAs in fluoride medium [22]. Unlike the polymorphs A and B, 150
the topology BEC has linear 12-ring channels and contains the d4r composite building unit. It has been shown that the presence of fluorine inside these units increases the stability. This can explain the preferential formation of the BEC phase in fluoride medium in our case. The polymorph C was previously obtained with different OSDAs [41, 46]. For example, a pure silica BEC-type zeolite 155
was prepared with 1,1’-(butane-1,4-diyl)bis(1-azabicyclo[2.2.2]octan-1-ium), but only as an overgrowth of β zeolite (ITQ-14 on Table2). The one in pure form was prepared with the OSDA 4,4-dimethyl-4-azoniatricyclo[5.2.2.02,6
]undec-8-ene [47]. Nevertheless, all the OSDAs used are significantly different from those used in our work. Thus, we demonstrated that the BEC-type zeolites can be 160
obtained with several new asymmetric OSDAs.
Most of the obtained phases contain d4r units (AST, UOZ, BEC, IWW, UOV, UOZ) which are favored by the presence of germanium and also by the presence of fluoride that is known to stabilize these units. Indeed, the only phases containing no d4r units are MFI and *BEA which never form pure 165
in a fluoride medium, which shows the importance of fluoride ions for the d4r stabilization.
It seems that most of our OSDAs tested are favorable to the formation of the large pore zeolites (12-ring) BEC, *BEA (β), IWW (ITQ-22) and UOV (IM-17) see Table3). Nevertheless, the influence of the medium seems to overcome 170
the effect of the OSDA in these syntheses. Our experiments demonstrate that the organic molecules mostly act as pore-fillers than real structure-directing agents. Indeed, the formation of BEC-type zeolites can often be observed with all the OSDAs as the main phase, if not as the unique phase (Table 6). Additionally, in section4below, the structure analysis also proves weak OSDA-175
framework interactions.
Pure BEC samples were thoroughly characterized in order to study the impact of media and OSDAs on the properties of the product.
3.2. Further characterizations from SEM, PXRD, TGA and EDX
Among the different germanosilicates, the polymorph C of zeolite beta (BEC) 180
is present as a dominant phase in five samples (see the corresponding XRD pat-terns in Figure S4 to S10). As it was mentioned above, the pure BEC phase is preferentially crystallized in a fluoride medium, and mainly in a concentrated gel. However, the sample 26 was obtained in the diluted one. A comparison of the obtained germanosilicates allows us to observe the differences between them 185
in spite of the synthesis in similar conditions. The SEM images of samples 5, 13 and 25 are shown in Figure S2. The zeolites are composed of rod-shaped crys-tals, but their size and the elongation vary considerably. A statistical study, carried out using SEM with data collected on fifty particles (Figure S2, S3 and Table S1), reveals that they have particles of a smaller size than 0.5µm 190
while the samples 9 and 26 (Figure 1) have rod-shaped crystals almost ten times larger. Comparing the samples 25 and 26 obtained with the same OSDA
Table 6: Products obtained from germanium-containing synthesis media (one week at 170◦C).
R(OH)2 Medium1 N◦ Products
TM[C5](C4N)
Conc.HF 1 AST
Dil.HF 2 BEC — UOZ — AST
Conc.OH 3 Amorphous — IWW
Dil.OH 4 Amorphous — IWW
TE[C5](C4N)
Conc.HF 5 BEC — *BEA
Dil.HF 6 Quartz (two types)
Conc.OH 7 *BEA — BEC
Dil.OH 8 Amorphous
TM[C6](C4N)
Conc.HF 9 BEC
Dil.HF 10 Quartz
Conc.OH 11 IWW — *BEA
Dil.OH 12 IWW
TE[C6](C4N)
Conc.HF 13 BEC
Dil.HF 14 BEC — MFI
Conc.OH 15 BEC — *BEA
Dil.OH 16 MFI — Amorphous
TM[C5](C5N)
Conc.HF 17 BEC — Argutite — AST
Dil.HF 18 BEC — Quartz
Conc.OH 19 UOV — AST
Dil.OH 20 UOV — MFI
TM[C6](C5N)
Conc.HF 21 BEC — Argutite Dil.HF 22 Argutite — BEC
Conc.OH 23 UOV — ?
Dil.OH 24 MFI
TM[C7](C5N)
Conc.HF 25 BEC
Dil.HF 26 BEC
Conc.OH 27 BEC — *BEA
Dil.OH 28 MFI
(TM[C7](C5N)), it seems that the diluted medium favors the formation of larger and more elongated crystals.
(a) (b)
Figure 1: SEM micrographs of BEC-type germanosilicate samples (a) 9 and (b) 26.
Thermogravimetric analyses were performed on these five samples (5, 9, 195
13, 25, 26) and compared with EDX results. The total weight loss including the water loss for temperatures under 200◦C, the template decomposition and the loss of fluorine coupled with EDX results led to the chemical formula of these compounds given in Table 7. The TGA curves (Figure S11) underline differences between these samples, in terms of occluded water, fluoride and 200
OSDA. Once again samples 9 and 26 behave differently, they contain only a very small amount of physisorbed water and almost no chemisorbed water is observed. The results observed by PXRD, SEM and TGA show that of the five samples of BEC, sample 9 is the purest and best crystallized.
Table 7: TGA and EDX measurement data with the corresponding chemical formulae for samples 9 and 26.
Sample Si/Ge H2O loss OSDA + F loss chemical formulaa
(EDX) (TGA)(%) (TGA)(%) (EDX + TGA)
9 2.1 0.9 17.1 |(C14H32N2)1.85F3.7|[Si21.7Ge10.3O64]
26 3.5 0.5 17.6 |(C16H36N2)1.62F3.24|[Si24.9Ge7.1O64] aThe fluoride content was adjusted to compensate the positive charges of the OSDAs.
Finally, regarding these additional characterizations, it is clear that two sam-205
ples stand out from the others : samples 9 and 26. However, when comparing the powder X-ray diffraction patterns of those two samples, it turns out that sample 9 with a smaller full width at half maximum of the Bragg peaks is a better candidate for a localization of the occluded OSDA through the Rietveld method.
210
3.3. 13C solid-state and liquid NMR spectroscopy
Samples 9 and 26 are formed in the presence of structure-directing agents that are different. The synthesis of sample 9 requires TM[C6](C4N) in a
con-centrated medium, whereas the sample 26 is obtained in the presence of a larger molecule TM[C7](C5N) and a diluted medium. In Figure 2 the liquid
215
13C NMR spectra of the aqueous solutions of both OSDAs are compared to
the solid-state NMR spectra performed on both samples. For sample 9 the OSDA (TM[C6](C4N)) is intact inside the pore (Figure 2a), but in the case of
sample 26 the signal around -44 ppm in Figure2bcorresponds to the occluded by-products resulting from a demethylation, also proved by liquid 1H NMR
220
spectroscopy (Figure S12).
(a) (b)
Figure 2:13C CP-MAS NMR spectra of BEC-type germanosilicate samples (a) 9 and (b) 26 showing their occluded organic templates compared with liquid NMR of the corresponding solutions.
4. Structure from Rietveld analysis
The PXRD pattern was indexed in a tetragonal primitive unit cell with the parameters a = 12.7361(8) ˚A, c = 13.2219(10) ˚A and V = 2144.7(3) ˚A3 by the Lou¨er’s indexing DICVOL91 [48] module of the STOE WinXP OW [49] 225
suite of programs.The observed systematic extinctions suggested the space group P 42/m m c as for ITQ-17 [41]. The Rietveld analysis was conducted with the
free software GSAS-II [50]. In a first step, a Le Bail refinement [51] allowed us to determine the background and profile parameters. Then, the atomic coordinates of the whole framework atoms of IM-17 (i.e. T and O atoms with T = Si or Ge) 230
were used as the starting model. Soft restraints were placed on the bond lengths and angles of the framework (T–O = 1.68(4) ˚A for T1 (germanium rich site) and 1.62(4) ˚A for T2-3 (silicon-rich sites) and O-T-O = 109.5(30)◦). The first stage of the Rietveld analysis was the scale factor determination. Initially, for the Rietveld refinement, on each T atom site, a silicon atom and a germanium atom 235
were both placed at the same position with the sum of their occupancy factors constrained to be 1. All the atoms were refined isotropically. After refinement of the framework, the three T atoms sites were mixed sites. Then the calculated Fourier difference maps revealed a scattering density inside the void volume attributed to the presence of the OSDA. The OSDA was introduced directly 240
inside the pore of this BEC through the program MCE [52]. Soft restraints (bonds and angles) were added in order to maintain a suitable geometry for the OSDA. At the end of the refinement, positive differences in intensities were observed at positions characteristic of the argutite ones. To account for this impurity, we added argutite [53] as a secondary phase.
245
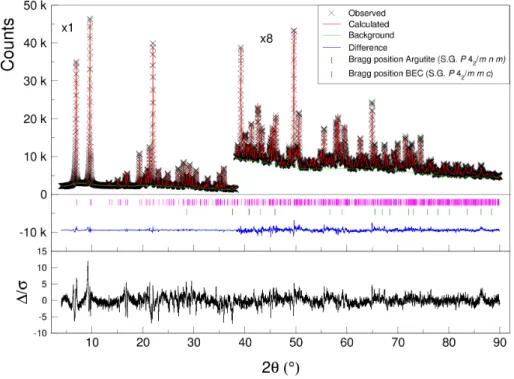
The final Rietveld refinement resulted in good reliability factors; the Ri-etveld plot is displayed in Figure3 and crystal as well as Rietveld refinement parameters are listed in Table 8. As for the atomic and isotropic displace-ment parameters, they can be seen in Table S2, respectively. Selected bond angles and distances are listed in Tables S3 and S4. CCDC 1998372 contains 250
the supplementary crystallographic data for this paper. The data can be ob-tained free of charge from The Cambridge Crystallographic Data Centre via
https://www.ccdc.cam.ac.uk/structures/.
Figure 3: The final Rietveld plot of the BEC-type germanosilicate, sample 9. The high angle part has been magnified by a factor of 8. ∆/σ is the weighted difference between the experimental and calculated diffraction patterns, the square of this weighted difference is what is actually minimized.
Table 8: Crystal and Rietveld refinement data for the BEC-germanosilicate (sample 9).a
Chemical formula per unit cell (refined) |(C14H32N2)1.90| [Si21.75Ge10.25O64F2] Space group P 42/m m c (#131)
λ (˚A), Cu Kα1 1.5406
Data collection temperature T (K) 293
a (˚A) 12.7320(3)
c (˚A) 13.2198(3)
V (˚A3) 2143.00(14)
Z 1
Number of data points, angular range (◦)(step (◦2θ)) 8600, 4.00-90.00 (0.01)
Number of contributing reflections 521 Number of profile parameters 7 Total number of refined parameters 211
Total number of restraints (bonds, angles) 174 (60, 114) Total number of constraints 15
bR p 0.02549 bwR p 0.03380 bR B 0.02531 bwR B 0.03071 bR exp 0.02235 bR F 0.03643 bR F2 0.05454 bGOF 1.564
Largest differences peak and hole (¯e/˚A3) 0.870 ; -0.667
aThe refinement gave a mass fraction of 0.0147(3) for the secondary phase argutite. bThe definition of these residual values are given in [50].
The asymmetric unit of the BEC-type germanosilicates (sample 9) is repre-255
sented in Figure4a. The occupancy factors of the T atoms (Table S2) illustrate a T1 site richer in germanium than T2 and T3, the refined Si/Ge molar ratio of 21.9/10.1 being similar to the one measured by EDX (21.7/10.3 in Table7). This was expected since all the T1 sites, after application of the symmetry oper-ations, are localized at the vertices of the d4r composite building unit, a typical 260
characteristic of microporous d4r -containing germanosilicates ([24,54,55,56]). The refinement reveals also that fluoride anions (F1) are localized at the center of the d4r units (Figure4b) with a full occupancy i.e., two fluoride anions per unit cell. This last point is confirmed by the solid-state MAS19F NMR spec-trum (Figure S13) where an asymmetric resonance is present at about -5 ppm 265
characteristic of F− occluded in d4r with the vertices partially substituted by
germanium. However, the shift of -5 ppm is surprising since the Rietveld re-finement resulted in an almost equal quantity of germanium and silicon atoms at the vertices of the d4r unit (see s.o.f. of T1 in Table S2), i.e. four silicon and germanium tetrahedra. From an experimental point of view, a chemical 270
shift of about -7 ppm was therefore expected for such d4r [57, 58, 59]. From a theoretical point of view, though, this chemical shift can be related to the work of Pulido et al. [60]. It concerns the calculation with a DFT methodol-ogy of the19F NMR chemical shift of fluoride ions occluded in d4r units. For
d4r units of composition Si5Ge3 and Si4Ge4 and depending on the number of 275
respect to CFCl3are -6, -2 and -1 ppm, respectively. In our case, the d4r units
are defined only by T1 sites, their refined average composition is Si4.056Ge3.944, which could, on the basis of these theoretical calculations explain a chemical shift of -5 ppm on the NMR spectrum. To our knowledge, this has never been 280
demonstrated experimentally until now. In addition, the spectrum exhibits a broad resonance ranging between -90 and -190 ppm with a global signal inten-sity slightly lower than the one at -5 ppm, reflecting a high disorder inside the pores. This explains why it was not possible to localize the extra-framework fluorides species. From the19F MAS NMR spectroscopy and the structure
pro-285
vided by Rietveld refinement, it is clear that the present fluoride ions (d4r -and non-localized ”free”-fluoride ions) compensate the positive charges brought about by the organic moities.
(a) (b)
Figure 4: (a) The asymmetric unit and (b) a perspective down [010] with a possible arrange-ment of two occluded organic molecules TM[C6](C4N) within the pores of one unit cell of the BEC-type germanosilicate (sample 9) as determined by the present Rietveld study (see Table S2).
There is a unique crystallographic site for the occluded OSDAs and, after the Rietveld refinement study, its site occupancy factor is 0.1181(5) or 1.9 OSDAs 290
per unit cell. In Figure4awe can also observe the deformation of the linear con-formation of the alkyl chain. The perspective view in Figure4bshows one unit cell with two OSDAs aligned inside the 12-ring channels along [100] and [010], respectively. With such an OSDA content and taking into account the previous characterizations and the refined Si/Ge molar ratio, the chemical formula for this 295
phase is |(C14H32N2)1.9F1.9| [Si21.75Ge10.25O64F2]. The corresponding fluoride and OSDA weight content is then of 17.5%, a percentage comparable to the one determined after the TGA and EDX measurements (see Table7). The hydrogen bonding scheme is represented in Figure5, the shorter contacts involve almost exclusively the methylpyrrolidinium group. The strongest interaction is between 300
not appropriate to give the OSDA a real role of a structuring-directing agent, it seems rather to have the role of a species serving as a pore filler. Such a result differs from the one obtained with the bis-imidazolium derivatives, which act as true OSDAs for the chiral silicogermanate of topology STW. Their specificity 305
towards this topology decreases with the increase in the length of the linker in a pure silica medium. However, in the presence of germanium, all the OSDAs lead to this topology which has only d4r as composite building units. Thus, no influence of the linker length is observed [11].
Figure 5: The hydrogen bonding scheme of the BEC-type germanosilicate, sample 9.
5. Conclusion 310
In summary, a series of syntheses using asymmetric diquaternary ammonium salts as OSDAs was performed in the (Si,Ge) system. We found out that the zeolite topology obtained through an hydrothermal synthesis depends greatly on the gel composition. Any tiny change can drastically influence the crystal-lization. Considering our results, trends stand out with regard to the OSDA 315
length and size, but the impact of the medium remains significant, especially the presence of fluoride and the dilution.
The seven OSDAs used in various conditions led to the crystallization of BEC-type zeolites. We observed that between BEC samples obtained with different organic molecules, morphological and chemical differences can occur. 320
Considering the presented syntheses of this zeolite, it appears that it is prefer-entially obtained in a pure state in fluoride and/or concentrated media. The absence of fluoride tends to favor multiphasic products, except in diluted media where IWW and MFI are obtained.
A structural study on one of the germanosilicates of topology BEC allowed 325
us to localize the organic molecule in the 12-ring channels, but its role as OSDA could not be proved due to rather weak interactions between the OSDA and the framework. Finally, even if the introduction of an asymmetry and the length of
the OSDA initially opened up the possibility to get different schemes of inter-actions between the framework and the guest molecules, the role of germanium 330
predominates in this type of synthesis due to its strong contribution to the for-mation of the d4r composite building units, which is enhanced with the presence of fluoride.
6. CRediT authorship contribution statement
Carole Isaac: Investigation, Writing. Jean-Louis Paillaud: Concep-335
tualization, Supervision, Investigation, Writing - Review & Editing. Andrey Ryzhikov: Supervision, Writing. Jean Daou: Supervision, Writing.
7. Acknowledgements
We thank the University of Upper Alsace (UHA) for a Doctoral grant to C. I. (Agreement No. 2018-61). We also thank S´everinne Rigolet from IS2M and 340
Didier Le Nou¨en from LIMA for their assistance with the solid-state NMR and liquid NMR experiments, respectively.
8. Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported 345
in this paper.
9. Appendix A. Supplementary data
Supplementary data related to this article can be found athttp://dx.doi. org/10.1016/j.micromeso.2020.xx.xxx.
References 350
[1] M. Guisnet, J.-P. Gilson, Introduction to zeolite science and technology, Catalytics Science Series, Imperial College Press, 2002, Ch. 1, pp. 1–28. [2] R. M. Milton, Molecular sieve adsorbents, US Patent 2,882,243 (April 14,
1959).
[3] R. M. Barrer, P. J. Denny, 201. Hydrothermal chemistry of the silicates.
355
Part IX. Nitrogenous aluminosilicates, J. Chem. Soc. (1961) 971–982.http: //dx.doi.org/10.1039/JR9610000971
[4] E. M. Flanigen, Chapter 2 Zeolites and molecular sieves: An historical perspective, in: H. van Bekkum, E. Flanigen, P. Jacobs, J. Jansen (Eds.), Introduction to Zeolite Science and Practice, Vol. 137 of Studies in Surface 360
[5] C. S. Cundy, P. A. Cox, The hydrothermal synthesis of zeolites: History and development from the earliest days to the present time, Chem. Rev. 103 (3) (2003) 663–702. https://doi.org/10.1021/cr020060i
[6] L. G´omez-Hortig¨uela, Insights into the chenistry of organic structure-365
directing agent in the synthesis of zeolitic materials, Vol. 175 of Structure and Bonding, Springer, 2010.
[7] R. L. Wadlinger, G. T. Kerr, E. J. Rosinski, Catalytic composition of a crystalline zeolite, US Patent 3,308,069 (March 7, 1967).
[8] R. Grosse, E. M. Flanigen, Crystalline silica, US Patent 4,061,724 (Decem-370
ber 6, 1977).
[9] P. Lu, L. G´omez-Hortig¨uela, Z. Gao, M. A. Camblor, Synthesis of a ger-manosilicate zeolite HPM-12 using a short imidazolium-based dication: structure-direction by charge-to-charge distance matching, Dalton Trans. 48 (2019) 17752–17762.http://dx.doi.org/10.1039/C9DT04089G
375
[10] M. O. Cichocka, Y. Lorgouilloux, S. Smeets, J. Su, W. Wan, P. Caullet, N. Bats, L. B. McCusker, J.-L. Paillaud, X. Zou,Multidimensional disorder in zeolite IM-18 revealed by combining transmission electron microscopy and x-ray powder diffraction analyses, Cryst. Growth Des. 18 (4) (2018) 2441–2451. https://doi.org/10.1021/acs.cgd.8b00078
380
[11] P. Lu, L. G´omez-Hortig¨uela, L. Xu, M. A. Camblor,Synthesis of STW zeo-lites using imidazolium-based dications of varying length, J. Mater. Chem. A 6 (2018) 1485–1495.http://dx.doi.org/10.1039/C7TA10002G
[12] D. Jo, S. B. Hong, Targeted synthesis of a zeolite with pre-established framework topology, Angew. Chem. Int. Ed. 58 (39) (2019) 13845–13848. 385
https://doi.org/10.1002/anie.201909336
[13] S.-H. Lee, C.-H. Shin, G. J. Choi, T.-J. Park, I.-S. Nam, B. Han, S. B. Hong,
Zeolite synthesis in the presence of flexible diquaternary alkylammonium ions (C2H5)3N+(CH2)nN+(C2H5)3 with n=3–10 as structure-directing agents, Microporous Mesoporous Mater. 60 (1) (2003) 237–249. http:
390
//www.sciencedirect.com/science/article/pii/S1387181103003810
[14] R. F. Lobo, S. I. Zones, R. C. Medrud,Synthesis and Rietveld refinement of the small-pore zeolite SSZ-16, Chem. Mater. 8 (10) (1996) 2409–2411.
https://doi.org/10.1021/cm960289c
[15] E. M. Flanigen, R. L. Patton, Silica polymorph and process for preparing 395
same, US Patent 4,073,865 (1978).
[16] J.-L. Guth, H. Kessler, R. Wey, New route to pentasil-type zeolites using a non alkaline medium in the presence of fluoride ions, in: Y. Murakami, A. Iijima, J. W. Ward (Eds.), Studies in Surface Science and Catalysis, Vol. 28, Elsevier, 1986, pp. 121–128.
[17] J.-L. Paillaud, P. Caullet, J. Brendl´e, A. Simon-Masseron, J. Patarin, The fluoride route: A good opportunity for the preparation of 2D and 3D in-organic microporous frameworks, in: A. Traissaud (Ed.), Functionalized Inorganic Fluorides: Synthesis, Characterization & Properties of Nanos-tructured Solids, John Wiley & Sons, Ltd, 2010, Ch. 16, pp. 489–518. 405
[18] P. Caullet, J. L. Guth, J. Hazm, J. M. Lamblin, H. Gies, Synthesis, characterization and crystal-structure of the new clathrasil phase octade-casil, Eur. J. Solid State Inorg. Chem. 28 (2) (1991) 345–361. http: //doi.wiley.com/10.1002/chin.199124028
[19] M. A. Camblor, A. Corma, P. Lightfoot, L. A. Villaescusa, P. A. Wright, 410
Synthesis and structure of ITQ-3, the first pure silica polymorph with a two-dimensional system of straight eight-ring channels, Angew. Chem. Int. Ed. 36 (23) (1997) 2659–2661. https://onlinelibrary.wiley.com/doi/ abs/10.1002/anie.199726591
[20] J.-L. Paillaud, Y. Lorgouilloux, B. Harbuzaru, P. Caullet, J. Patarin, 415
N. Bats, Structure orienting role of germanium in zeolite synthesis, in: Studies in Surface Science and Catalysis, Vol. 170, Elsevier, 2007, pp. 389– 396.
[21] C. S. Cundy, P. A. Cox,The hydrothermal synthesis of zeolites: Precursors, intermediates and reaction mechanism, Micropororous Mesopororous Mat. 420
82 (1-2) (2005) 1–78.https://doi.org/10.1016/j.micromeso.2005.02. 016
[22] T. Conradsson, M. Dadachov, X. Zou, Synthesis and structure of (Me3N)6[Ge32O64].(H2O)4.5, a thermally stable novel zeotype with 3D interconnected 12-ring channels, Micropororous Mesopororous Mat. 41 425
(2000) 183–191.https://doi.org/10.1016/S1387-1811(00)00288-2
[23] Y. Mathieu, J.-L. Paillaud, P. Caullet, N. Bats, Synthesis and charac-terization of IM-10: A new microporous silicogermanate with a novel topology, Micropororous Mesopororous Mat. 75 (1) (2004) 13–22.https: //doi.org/10.1016/j.micromeso.2004.06.023
430
[24] Y. Lorgouilloux, M. Dodin, E. Mugnaioli, C. Marichal, N. Bats, P. Caul-let, U. Kolb, J.-L. Paillaud, IM-17: a new zeolitic material, synthesis and structure elucidation from electron diffraction ADT data and Rietveld analysis, RSC Adv. 4 (2014) 19440–19449.http://dx.doi.org/10.1039/ C4RA01383B
435
[25] A. Rojas, L. G´omez-Hortig¨uela, M. A. Camblor,Zeolite structure direction by simple bis(methylimidazolium) cations: The effect of the spacer length on structure direction and of the imidazolium ring orientation on the 19F NMR resonances, J. Amer. Chem. Soc. 134 (8) (2012) 3845–3856.https: //doi.org/10.1021/ja210703y
[26] P. Caullet, J.-L. Paillaud, Y. Mathieu, N. Bats, Synthesis of zeolites in the presence of diquaternary alkylammonium ions as structure-directing agents, Oil Gas Sci. and Technol. 62 (6) (2007) 819–825. https://doi. org/10.2516/ogst:2007079
[27] A. Moini, K. Schmitt, E. Valyocsik, R. Polomski,The role of diquaternary
445
cations as directing agents in zeolite synthesis, Zeolites 14 (7) (1994) 504 – 511.https://doi.org/10.1016/0144-2449(94)90182-1
[28] N. Bats, L. Rouleau, J.-L. Paillaud, P. Caullet, Y. Mathieu, S. Lacombe, Recent developments in the use of hexamethonium salts as structure direct-ing agents in zeolite synthesis, in: E. VanSteen, M. Claeys, L. H. Callanan 450
(Eds.), Recent Advances in the Science and Technology of Zeolites and Re-lated Materials, Part A, Vol. 154 of Studies in Surface Science and Catal-ysis, Elsevier Science Bv, Amsterdam, 2004, pp. 283–288.
[29] A. Corma, M. Puche, F. Rey, G. Sankar, S. J. Teat, A zeolite structure (ITQ-13) with three sets of medium-pore crossing channels formed by
9-455
and 10-rings, Angew. Chem. Int. Ed. 42 (10) (2003) 1156–1159. https: //doi.org/10.1002/anie.200390304
[30] X. Liu, U. Ravon, A. Tuel,Effect of HF concentration on the composition and distribution of Ge species in the framework of ITQ-13 and ITQ-17 zeolites, Micropororous Mesopororous Mat. 170 (2013) 194–199. https:
460
//doi.org/10.1016/j.micromeso.2012.11.038
[31] M. D. Shannon, J. L. Casci, P. A. Cox, S. J. Andrews, Structure of the two-dimensional medium-pore high-silica zeolite NU-87, Nature 353 (6343) (1991) 417–420.https://doi.org/10.1038/353417a0
[32] F. Gramm, C. Baerlocher, L. B. McCusker, S. J. Warrender, P. A. Wright, 465
B. Han, S. B. Hong, Z. Liu, T. Ohsuna, O. Terasaki, Complex zeolite structure solved by combining powder diffraction and electron microscopy, Nature 444 (7115) (2006) 79–81.https://doi.org/10.1038/nature05200
[33] S. B. Hong, E. G. Lear, P. A. Wright, W. Zhou, P. A. Cox, C.-H. Shin, J.-H. Park, I.-S. Nam,Synthesis, structure solution, characterization, and
470
catalytic properties of TNU-10: A high-silica zeolite with the STI topology, J. Amer. Chem. Soc. 126 (18) (2004) 5817–5826. https://doi.org/10. 1021/ja031981t
[34] E. Benazzi, J.-L. Guth, L. Rouleau, IM-5 zeolite, a process for its prepara-tion and catalytic applicaprepara-tions thereof, US Patent 6,136,290 (October 21, 475
1997).
[35] C. Baerlocher, F. Gramm, L. Mass¨uger, L. B. McCusker, Z. He, S. Hovm¨oller, X. Zou,Structure of the polycrystalline zeolite catalyst IM-5 solved by enhanced charge flipping, Science 315 (5815) (2007) 1113–1116.
https://science.sciencemag.org/content/315/5815/1113
[36] C. Baerlocher, D. Xie, L. B. McCusker, S.-J. Hwang, I. Y. Chan, K. Ong, A. W. Burton, S. I. Zones, Ordered silicon vacancies in the framework structure of the zeolite catalyst SSZ-74, Nat. Mater. 7 (8) (2008) 631–635.
https://doi.org/10.1038/nmat2228
[37] P. Guo, K. Strohmaier, H. Vroman, M. Afeworki, P. I. Ravikovitch, C. S. 485
Paur, J. Sun, A. Burton, X. Zou, Accurate structure determination of a borosilicate zeolite EMM-26 with two-dimensional 10 × 10 ring channels using rotation electron diffraction, Inorg. Chem. Front. 3 (2016) 1444–1448.
http://dx.doi.org/10.1039/C6QI00262E
[38] A. F´ecand, N. Bats, Crystalised solid IZM-3 and method for preparing 490
same, US Patent 8,361,427 B2 (2007).
[39] Y. Li, D. Bernin, F. Gao, N. Hedin,Microporous pure-silica IZM-2, Micro-pororous MesoMicro-pororous Mat. 237 (2017) 222–227. https://doi.org/10. 1016/j.micromeso.2016.09.033
[40] A. F´ecand, N. Bats, IZM-2 crystalline solid and process for its preparation, 495
US Patent 8,361,435 B2 (2007).
[41] A. Corma, M. T. Navarro, F. Rey, J. Rius, S. Valencia, Pure Polymorph C of Zeolite Beta Synthesized by Using Framework Isomorphous Substitu-tion as a Structure-Directing Mechanism, Angew. Chem. Int. Ed. 40 (12) (2001) 2277–2280. https://doi.org/10.1002/1521-3773(20010618)40:
500
12%3C2277::AID-ANIE2277%3E3.0.CO;2-O
[42] Z. Liu, T. Ohsuna, O. Terasaki, M. A. Camblor, M.-J. Diaz-Caba˜nas, K. Hi-raga, The first zeolite with three-dimensional intersecting straight-channel system of 12-membered rings, J. Am. Chem. Soc. 123 (22) (2001) 5370– 5371.https://doi.org/10.1021/ja0107778
505
[43] H. Li, O. M. Yaghi,Transformation of germanium dioxide to microporous germanate 4-connected nets, J. Am. Chem. Soc. 120 (40) (1998) 10569– 10570.https://doi.org/10.1021/ja982384n
[44] T. De Baerdemaeker, B. Yilmaz, U. M¨uller, M. Feyen, F.-S. Xiao, W. Zhang, T. Tatsumi, H. Gies, X. Bao, D. De Vos, Catalytic
appli-510
cations of OSDA-free Beta zeolite, J. Catal. 308 (2013) 73–81. https: //doi.org/10.1016/j.jcat.2013.05.025
[45] J. Higgins, R. LaPierre, J. Schlenker, A. Rohrman, J. Wood, G. Kerr, W. Rohrbaugh, The framework topology of zeolite beta, Zeolites 8 (6) (1988) 446–452.https://doi.org/10.1016/S0144-2449(88)80219-7
515
[46] A. Burton, Recent trends in the synthesis of high-silica zeolites, Catal. Rev. 60 (1) (2018) 132–175.https://doi.org/10.1080/01614940.2017. 1389112
[47] A. Cant´ın, A. Corma, M. D´ıaz-Caba˜nas, J. L. Jord´a, M. Moliner, F. Rey,
Synthesis and characterization of the all-silica pure polymorph C and an
520
enriched polymorph B intergrowth of zeolite beta, Angew. Chem. Int. Ed. 45 (47) (2006) 8013–8015. https://doi.org/10.1002/anie.200603027
[48] A. Boultif, D. Lou¨er, Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method, J. Appl. Crystallogr. 24 (6) (1991) 987–993. https://doi.org/10.1107/
525
S0021889891006441
[49] STOE & Cie GmbH, WinXP OW, version 2.20, Darmstadt, Germany, 12
June 2006.
[50] B. H. Toby, R. B. Von Dreele, GSAS-II : the genesis of a modern open-source all purpose crystallography software package, J. Appl. Crystallogr. 530
46 (2) (2013) 544–549.https://doi.org/10.1107/S0021889813003531
[51] A. Le Bail, H. Duroy, J. L. Fourquet,Ab-initio structure determination of LiSbWO6 by x-ray powder diffraction, Mater. Res. 23 (3) (1988) 447–452. https://doi.org/10.1016/0025-5408(88)90019-0
[52] J. Rohl´ıˇcek, M. Huˇs´ak, MCE2005 – a new version of a program for fast
535
interactive visualization of electron and similar density maps optimized for small molecules, J. Appl. Crystallogr. 40 (3) (2007) 600–601. https: //doi.org/10.1107/S0021889807018894
[53] W. H. Baur,Uber die Verfeinerung der Kristallstrukturbestimmung einiger¨ Vertreter des Rutiltyps: TiO2, SnO2, GeO2 und MgF2, Acta Crystallogr.
540
9 (6) (1956) 515–520. https://doi.org/10.1107/S0365110X56001388
[54] A. Corma, F. Rey, S. Valencia, J. L. Jord´a, J. Rius,A zeolite with intercon-nected 8-, 10- and 12-ring pores and its unique catalytic selectivity, Nat. Mater. 2 (7) (2003) 493–497.https://doi.org/10.1038/nmat921
[55] J.-L. Paillaud, B. Harbuzaru, J. Patarin, N. Bats, Extra-large-pore
zeo-545
lites with two-dimensional channels formed by 14 and 12 rings, Science 304 (5673) (2004) 990–992.https://science.sciencemag.org/content/ 304/5673/990
[56] M. Moliner, T. Willhammar, W. Wan, J. Gonz´alez, F. Rey, J. L. Jorda, X. Zou, A. Corma, Synthesis design and structure of a multipore zeolite
550
with interconnected 12- and 10-MR channels, J. Am. Chem. Soc. 134 (14) (2012) 6473–6478.https://doi.org/10.1021/ja301082n
[57] T. Blasco, A. Corma, M. D´ıaz-Caba˜nas, F. Rey, J. A. Vidal-Moya, C. M. Zicovich-Wilson, Preferential location of Ge in the double four-membered ring units of ITQ-7 zeolite, J. Phys. Chem. B 106 (10) (2002) 2634–2642. 555
[58] Y. Wang, J. Song, H. Gies, The substitution of germanium for silicon in AST-type zeolite, Solid State Sci. 5 (11) (2003) 1421–1433.https://doi. org/10.1016/j.solidstatesciences.2003.09.003
[59] G. Sastre, J. A. Vidal-Moya, T. Blasco, J. Rius, J. L. Jord´a, M. T. Navarro, 560
F. Rey, A. Corma, Preferential location of Ge atoms in polymorph C of Beta zeolite (ITQ-17) and their structure-directing effect: A compu-tational, XRD, and NMR spectroscopic study, Angew. Chem. Int. Ed. 41 (24) (2002) 4722–4726.https://onlinelibrary.wiley.com/doi/abs/ 10.1002/anie.200290028
565
[60] A. Pulido, G. Sastre, A. Corma,Computational study of 19F NMR spec-tra of double four ring-containing Si/Ge-zeolites, ChemPhysChem 7 (5) (2006) 1092–1099. https://chemistry-europe.onlinelibrary.wiley. com/doi/abs/10.1002/cphc.200500634
Highlights
570
• Hydrothermal synthesis of germanosilicate with 7 asymmetric diquarte-nary ammonium salts.
• Influence of the composition of the starting gel on the crystallization step. • Localization of the guest molecule in the pore volume by Rietveld. • Unusual deshielding on the19F MAS NMR spectrum of F−located in the
575
d4r CBUs.
![Table 1: Some symmetric diazolium-based imidazolium [I.] and (pyrazolium [P.] deriva- deriva-tives) salts used as OSDAs in zeolite synthesis.](https://thumb-eu.123doks.com/thumbv2/123doknet/14700290.564567/5.918.208.712.272.857/table-symmetric-diazolium-imidazolium-pyrazolium-deriva-zeolite-synthesis.webp)