Complex Mechanical Design of Bio-Inspired Model
Transient Network Hydrogels
by
Scott C. Grindy
B.S., Northwestern University (2012)
Submitted to the Department of Materials Science and Engineering
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Materials Science and Engineering
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY
June 2017
© Massachusetts Institute of Technology 2017. All rights reserved.
Author . . . .
Department of Materials Science and Engineering
May 12, 2017
Certified by . . . .
Niels Holten-Andersen
Assistant Professor
Thesis Supervisor
Accepted by . . . .
Donald R. Sadoway
Chairman, Departmental Committee on Graduate Studies
Complex Mechanical Design of Bio-Inspired Model Transient
Network Hydrogels
by
Scott C. Grindy
Submitted to the Department of Materials Science and Engineering on May 12, 2017, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy in Materials Science and Engineering
Abstract
The mechanical properties of viscoelastic soft materials are strongly time-dependent, such that we must describe their mechanical properties with material functions. This is an inherently difficult problem for materials scientists: typically, we define structure-property relationships in terms of scalar material properties, such that modifying a ma-terial’s structure affects a target material property. However, if the property of interest is function-valued, modifying the material’s structure may affect different parts of the material function in undesirable ways. The increased dimensionality of the target ma-terial property therefore renders the overall mama-terials design problem for soft mama-terials significantly more difficult.
Recently, transient interactions have been shown to vastly improve the mechani-cal properties of soft materials by providing increased energy dissipation through the dissociation of the reversible bonds. However, there is a wide variety of transient in-teractions to choose from, varying widely in binding strength, kinetics, specificity, and stoichiometry of the groups that form the association. More research needs to be done to identify what physical laws apply universally across the types of transient associa-tions, and what differentiates the abilities of different types of interactions to control material mechanics.
In this thesis, we show how transient metal-coordinate bonds inspired by the chem-istry of the mussel byssal threads can be used to engineer viscoelastic material func-tions in an intuitive and facile manner. We show that intelligent understanding of the thermodynamics and kinetics of metal-coordinate complexes allows quasi-independent control over different regimes of the viscoelastic material function. We draw from clas-sical polymer physics and metal-coordinate chemistry to show that our 4-arm polyethy-lene glycol-based hydrogels crosslinked with transient histidine:metal bonds repre-sent a uniquely ideal system for probing fundamental questions in how the proper-ties of transient interactions affect viscoelastic material functions. In the final part of
this thesis, we extend our control over the viscoelastic material functions of hydrogels by exploiting the redox-sensitivity of histidine:metal crosslinks. In this way, we show how histidine:metal interactions are perhaps more versatile than other types of tran-sient interactions, promising a facile way to examine structure-property relationships in transient networks.
Thesis Supervisor: Niels Holten-Andersen Title: Assistant Professor
Acknowledgments
First and foremost, I would like to thank my advisor Niels Holten-Andersen for sup-porting me throughout the last five years. Niels took a huge chance by taking on a student whose only research experience was in computational crystalline materials to do research on experimental soft materials. I’ve always loved learning new things, and Niels only encouraged me to push my boundaries even further. I had a ton of intel-lectual freedom in grad school, and I have Niels to thank for allowing me to indulge in some hare-brained ideas that lead to fruitful discoveries. I’ve often said that Niels is the nicest person I know, and without his advising my research career would not be the same.
I’d also like to acknowledge my committee, Professors Alfredo Alexander-Katz and Krystyn Van Vliet. Their experience and advice over the years was incredibly vital to me. As a brand-new research group, much of our work was figuring out which scientific niche we should occupy, and their advice was instrumental in helping us locate that niche.
I had several other mentors during my PhD studies whose guidance was key to my formative years as a scientist. Reid Van Lehn and Michelle Sing-Ehret were particularly generous with their time and advice teaching me both how to be a scientist and answer-ing my questions about basic polymer science and soft matter physics. I’m grateful to be able to call them mentors, friends, and colleagues.
On the personal level, I’d like to acknowledge my fellow DMSE friends for making my time at MIT much more fun and enriching. In particular, Michael Campion, Chris Heidelberger, and Steve Dacek for being awesome roommates and always being up for esoteric discussions of materials science. I also want to thank my parents Cindy and Steve and brother Mark for support in various ways over the years; in particular my mom for fostering my interest in science since practically birth. Lastly, I want to thank my girlfriend Maureen for all her love, support, encouragement, and for always picking me up when I was down. Her contribution to this thesis may not be written on its pages,
but I can’t thank her enough.
Finally, I’d like to thank several institutions for funding my research: the Depart-ment of Materials Science and Engineering at MIT through the Anne M. Mayes Fel-lowship, the U.S. National Science Foundation under award number DMR - 1419807, the U.S. Office of Naval Research under the Young Investigators Program Grant ONR. N00014-15-1-2763, and by a grant from 3M Company.
Contents
1 Introduction 19
1.1 Scalar-Valued vs. Function-Valued Material Properties . . . 19
1.2 Hydrogels and Transient Networks . . . 21
1.3 Mussel Byssi and Metal-Coordination . . . 22
1.4 Overview of the rest of the thesis . . . 23
1.5 Works not discussed in detail in this thesis . . . 26
2 Background 29 2.1 Time-Dependent Mechanical Properties: Viscoelasticity and Rheology . 29 2.1.1 Mechanical Tests for Viscoelastic Materials . . . 31
2.1.2 Phenomenological Models for Viscoelastic Materials . . . 33
2.2 Supramolecular Polymeric Hydrogels . . . 36
2.2.1 The Theory of Transient Networks . . . 38
2.2.2 Kinetically-Controlled Supramolecular Materials . . . 39
2.3 Metal-coordinate chemistry . . . 40
2.3.1 Metal-Coordination Thermodynamics and Kinetics . . . 40
2.4 Metallosupramolecular Network Mechanics . . . 43
2.5 Metal-Coordination in Biology and Bio-inspired Materials: The Mussel Byssus Thread . . . 45
2.5.1 Metal-Coordination in the byssus . . . 45
3 Methods 51
3.1 Synthesis of 4PEG-His . . . 51
3.2 Synthesis of 4PEG-DOPA (from D. Barrett) . . . . 51
3.3 NaOH-Controlled Synthesis of 4PEG Hydrogels . . . 52
3.4 Buffer-Controlled Synthesis of 4PEG Hydrogels . . . 53
3.5 Rheometric Measurements of 4PEG-His:M2+and 4PEG-His : 4PEG-dopa : M2+ . . . 53
3.6 Synthesis of 4PEG-NHS:4PEG-NH2:2+Hydrogels . . . 53
3.7 Rheometric Measurements of 4PEG-NHS:4PEG-NH2 : 4PEG-His : M2+ Hydrogels . . . 54
3.8 Synthesis of Linear Imidazole-Containing Polymer Melts . . . 54
3.9 Rheometric Measurements of L-ICP Melts . . . 55
3.10 Calculating the Relaxation SpectrumH(τ) using the double-lognormal assumption . . . 56
3.11 The Newton-Raphson Method . . . 57
3.12 Synthesis of LAP photoinitiator: . . . 57
4 Controlling Hierarchical Relaxation in Hydrogels 61 5 Linear and Non-Linear Viscoelasticity of 4PEG-His Hydrogels 77 5.1 Introduction . . . 77
5.2 Results and Discussion . . . 78
5.2.1 Polymer concentration and molecular weight primarily control elasticity . . . 78
5.2.2 The Metal/ligand ratio defines a window of highly tunable stiff-ness and relaxation time . . . 83
5.3 Nonlinear Properties . . . 88
6 Competitive Ligands and Exchange Processes 97
6.1 Introduction . . . 97
6.2 Double-metal Rheology . . . 99
6.2.1 Percolation affects only the slow relaxation mode . . . 102
6.2.2 Free ligands strongly affect the relaxation timescale by shorten-ing the effective lifetime of a bond . . . 106
6.3 The competitive ligand phenomena shows that weak ligands are an op-portunity for viscoelastic materials design . . . 113
6.4 Addendum . . . 116
7 UV-programmable viscoelastic material functions 119 7.1 Introduction . . . 119
7.2 Results and Discussion . . . 122
7.2.1 Single-Metal Gels . . . 122
7.2.2 Double-metal hydrogels . . . 126
7.3 Conclusions . . . 133
7.4 Methods specific to this chapter . . . 135
8 Conclusions & Future Outlook 137 8.1 Summary . . . 137
8.2 Future Directions and Open Questions . . . 141
8.2.1 His:Metal bonds and mechanics . . . 141
8.2.2 Photo-triggered Co3+bonds . . . 142
A Estimation of Relaxation Spectra through Bayesian Monte Carlo-based Sta-tistical Inference 145 A.1 Background . . . 145
A.2 Implementation . . . 150
B Code & Documentation 161
B.1 Log-normal relaxation spectra code . . . 161
B.2 Time-Temperature Superposition . . . 166
B.3 The Miller-Macosko Method for calculating percolation . . . 169
List of Figures
1.1 The mussel byssus . . . . 24
2.1 “A Rheological Chart” . . . 30
2.2 Mechanical tests of soft materials . . . 32
2.3 Constitutive models for viscoelastic materials. . . 34
2.4 The canonical model for viscoelastic materials and the relaxation spec-trum are derived from arrays of parallel-aligned Maxwell elements. . . . 35
2.5 Oscillatory deformation of the Maxwell model . . . 37
2.6 Equilibrium constants for a series of ligands and metals, showing the Irving-Williams series relationship. . . 41
2.7 Ligand exchange rate constants for several ligands and the relevant metal ions studied in this thesis. . . 42
2.8 The structure of Pre-Col proteins which compose the thread core. . . . 47
2.9 Growth in the number of peer-reviewed papers on the topic of mussel-inspired materials. . . 48
3.1 NMR of 4PEG-His precursor and samples at different molecular weights. 58 3.2 Synthesis of L-ICP polymer using RAFT polymerization. . . 59
4.1 Model materials systems with orthogonally tunable mechanical tem-poral hierarchy. . . 64
4.3 Hierarchical mechanics of double metal-ion coordinate networks. . . . 69 4.4 Relative contributions of slow and fast relaxation modes in double
metal-ion hydrogels. . . 70 4.5 The effects of temporal mechanical hierarchy in systems beyond model
4PEG-His hydrogels. . . 74
5.1 4PEG-His:Ni model system . . . 79 5.2 Linear rheology of 4PEG-His:Ni2+hydrogels at varying φ andMn . . . . 80 5.3 φ- andMn-scaling ofGP and τ . . . 81 5.4 Oscillatory rheology of 4PEG-His:Ni2+hydrogels at varying Ni2+/His ratio. 84 5.5 Scaling ofGP and τ with varying Ni2+/His ratio . . . 86 5.6 Crosslink geometry & elastically active chains . . . 87 5.7 Schematic of quantities used to describe large-amplitude oscillatory shear
experimental data (LAOS). . . 89 5.8 Large-amplitude oscillatory shear experimental data of 4PEG-His:Ni
hy-drogels. . . 92
6.1 In multiple-metal hydrogels with kinetically distinct exchange kinetics, slow relaxation modes are faster than their single-metal counterparts, while fast relaxation modes are slower than their single-metal counter-parts. . . 99 6.2 In supramolecular networks with kinetically distinct supramolecular
cross-links, two distinct relaxation modes exist. . . 100 6.3 The fast and slow relaxation modes of 4PEG-His:Ni,Cu networks have
similar trends as those in 4PEG-His:Ni,Co networks. . . 101 6.4 The fast and slow relaxation modes of 4PEG-His:Ni:Cu networks have
6.5 Comparison of theoretical moduli accounting for percolation affects with experimental measurements of the modulus associated with re-laxation from His:Ni crosslinks. . . 107 6.6 The effective number of free ligands . At the slow timescale, there is an
increase in the effective number of free ligands because the fast cross-links act as free ligands. At the fast timescale, there effective number of free ligands decreases. . . 109 6.7 Free ligands used to probe bond renormalization effects on hydrogels. . 111 6.8 Adding secondary ligands increases the expected plateau modulus and
the concentration of free ligands. . . 112 6.9 Increasing the open ligand concentration through the addition of
sec-ondary competitive ligands monotonically decreases the relaxation time. 113 6.10 We can exploit the effect of weakly-coordinating ligands to engineer
vis-coelastic material functions. . . 115 6.11 Relaxation spectra of 4PEG-His:Ni,Co hydrogels using the Bayesian
frame-work developed in Appendix A . . . 116 6.12 Relaxation spectra of 4PEG-His:Ni,Cu hydrogels using the Bayesian
frame-work developed in Appendix A . . . 117 7.1 Schematic of using photo-generated radicals to modify the viscoelastic
material functions of 4PEG-His:M hydrogels. . . 121 7.2 The LAP photoinitiator curing kinetics are significantly faster than the
commercially-available alternative, Irgacure 2959. . . 121 7.3 The radicals generated by the photodissociation of LAP react with the
His:M2+crosslinks in different ways depending on the metal ion, result-ing in a variety of viscoelastic properties which can be triggered by low-intensity UV irradiation. . . 123 7.4 Frequency sweeps of 4PEG-His:(Ni, Cu, or Co) hydrogels after UV
7.5 Comparison of UV-Vis absorption between 4PEG-His:Cu after treatment with photo-generated radicals or with ascorbic acid. . . 125 7.6 Color shifts of 4PEG-His:Co + LAP hydrogels upon exposure to UV light
and stability against chelating and reducing agents. . . 127 7.7 Using combinations of metal ions results in hydrogels with multiple
characteristic energy dissipation timescales. . . 128 7.8 Viscoelastic properties of 4PEG-His:Ni,Cu hydrogels both before and
af-ter UV irradiation. . . 129 7.9 Viscosity of 4PEG-His:Ni,Cu hydrogels after UV-irradiation. . . 130 7.10 Viscoelastic properties of 4PEG-His:Ni,Co hydrogels both before and
af-ter UV irradiation. . . 131 7.11 Viscoelastic properties of 4PEG-His:Co,Cu hydrogels both before and
after UV irradiation. . . 133 7.12 UV-rheometer setup. . . 135
8.1 Target (blue) and initial experimental moduli (red) for design problem. . 138
A.1 Example moduli used for demonstrating Monte Carlo estimation of re-laxation spectra. . . 151 A.2 Trace from example Monte Carlo estimation. . . 152 A.3 Inferred relaxation spectra and reconstructed moduli, showing the
ar-ray of possible sets ofgi that are consistent with our observed data. . . . 153 A.4 Inferred relaxation spectra from 4PEG-His:Ni,Co hydrogels with His:Mtotal
= 2:1. . . 156 A.5 Inferred relaxation spectra from 4PEG-His:Ni,Co hydrogels with His:Mtotal
= 3:1. . . 157 A.6 Inferred relaxation spectra from 4PEG-His:Ni,Cu hydrogels with His:Mtotal
A.7 Inferred relaxation spectra from 4PEG-His:Ni,Cu hydrogels with His:Mtotal
= 3:1. . . 159 A.8 Pearson’s correlation coefficient for the modulusgiassociated with each
discrete relaxation mode at τi from the Monte Carlo trace. . . 160 B.1 Example of time-temperature superposition. . . 166 B.2 Connecting the terminology of Miller and Macosko [129] to our
4PEG-His:M system. . . 169 B.3 Example of Evans et al. conversion of creep modulus to dynamic moduli 177
List of Tables
6.1 Time-dependent percolation: in networks with multiple distinct cross-link species, we must consider percolation at three distinct timescales. . 104 6.2 Species and stability constants used in equilibria calculations for metal
ions with histamine as a proxy for the His groups of 4PEG-His. . . 118 7.1 Summary of pre-UV and post-UV energy dissipation timescales of His:Ni,
Chapter 1
Introduction
1.1
Scalar-Valued vs. Function-Valued Material
Proper-ties
Materials scientists quantitatively design and evaluate materials with intrinsic mea-sures of a material’s performance which are normalized to the size of the material. Resistance becomes resistivity, and stiffness becomes Young’s modulus. We call these measures of material performance material properties. Most conventional material properties are scalar-valued. For example, the Young’s modulus of a material is 50 megapascals or its resistivity is 10 Ohm-meters.
Many material properties, by contrast, are function-valued - we can’t just write down a single number that describes the material’s behavior. Rather, the material prop-erty is dependent on an external input. An example of a material function is the ab-sorptivity of a solution ε(λ), where at each wavelength of light, the material absorbs a different fraction of the incident light. Because ε varies with an experimental condi-tion (the wavelength of light), the absorptivity is a funccondi-tion-valued materials property. Often, researchers approximate material functions either by discretizing them (record-ing the absorptivity at a certain wavelength or set of wavelengths) or by approximat(record-ing
CHAPTER 1. INTRODUCTION
them with a closed-form expression which describes (most of ) the observed material function. When we approximate the material property, we reduce its dimensionality for the sake of reducing its complexity, but we sacrifice the richness of information contained in the material function.
Function-valued material properties are fundamentally more difficult to set as a design target than scalar-valued properties explicitly because function-valued prop-erties have many more degrees of freedom. Therefore, when evaluating a material’s mechanical performance, we tend to use scalar-valued properties such as the Young’s modulus, yield strength, or extensibility (strain to failure) for solid materials and the viscosity for fluid materials. However, many materials are neither perfectly solid nor perfectly fluid, and one particular class of these materials are called viscoelastic. Most soft materials such as polymers, gels, colloids, and pastes fall into this category.
The load-deformation relationship for soft materials strongly depends on the ef-fective timescale of the applied deformation, so viscoelastic materials can only be suf-ficiently described mechanically by function-valued material properties. Rather than using the scalar shear modulusG as we would for a solid material, viscoelastic mate-rials are described by the time-dependent stress relaxation modulusG(t) (or equiva-lently the frequency-dependent complex shear modulusG∗(ω)), even within the linear (low-strain) regime. Therefore, when engineering soft, viscoelastic materials, material property design targets should be function-valued.
A typical mechanical design problem for a soft material then goes as follows: first, quantify the specific material function that optimizes system performance, then iden-tify the material system that has that specific mechanical material function. Both of these steps are non-trivial; while the first step is independent of the material itself, the complexity of optimizing over a high-dimensional material function space is diffi-cult [1, 2], and the second step is severely limited by the requirements on the materi-als chemistry available. Therefore, design strategies targeted towards function-valued material properties are still in their infancy.
CHAPTER 1. INTRODUCTION
In this thesis, we seek to address the second challenge: creating materials with “de-signer” function-valued material properties. Our approach uses a model system of PEG-based hydrogels crosslinked with bio-inspired histidine-transition metal cross-links; by mildly modifying the materials chemistry we are able to dramatically alter one particular function-valued material property, the viscoelastic modulusG∗(ω). In doing so we identify and investigate general materials chemistry strategies for designing the frequency-dependent complex modulus of soft materials.
1.2
Hydrogels and Transient Networks
Hydrogels are a promising class of soft materials for use in bio-functional applications. Due to their relatively high water content (usually ∼70-99% by mass) and primarily organic makeup, the basic structure of hydrogels resembles that of living tissue, and hydrogels have found applications both in vivo as synthetic implantable tissues, ad-hesives, drug delivery devices, or contact lenses and in vitro as cell-culture materi-als. Super-absorbent polymers, used to clean up spills and in diapers, are also types of hydrogels. While hydrogels have found many applications across various settings, hydrogels’ performance is typically limited by their poor mechanical properties.
Most common hydrogels are made by randomly crosslinking polymer chains in an aqueous solution, giving the final hydrogel an essentially randomly-connected net-work structure. Within this basic structure, the properties of the hydrogel can be tuned by simply varying the monomer chemistry, crosslink density, or polymer concentra-tion, but the random crosslink structure is often the primary cause of their poor me-chanical performance.
In order to overcome this mechanical limitation, researchers incorporate transient, reversible interactions into hydrogels’ structure. Using transient interactions either alongside or in place of covalent crosslinks have been shown to be a promising strat-egy for engineering hydrogel materials with improved mechanical properties [3]. While
CHAPTER 1. INTRODUCTION
covalent crosslinks are permanent and fixed in place, transient interactions are by def-inition reversible and transient bonds are constantly breaking and re-forming, effec-tively rearranging the polymer network structure. Hydrogels which incorporate tran-sient bonds tend to be more durable than their permanently-crosslinked counterparts because they can respond to external loading or deformation by rearranging the net-work structure rather than break the structure irreversibly. Netnet-works of polymer chains crosslinked by reversible interactions are called supramolecular networks.
In this thesis, we employ bio-inspired metal-coordinate transient crosslinks to en-gineer the (viscoelastic) mechanical properties of supramolecular hydrogels. Our spe-cific aims are to (1) elucidate and investigate the mechanisms of relaxation and (2) us-ing this understandus-ing, demonstrate how these types of bonds differentiate themselves from other transient interactions through their unique sensitivity to their local bonding environment.
1.3
Mussel Byssi and Metal-Coordination: Inspiration for
the design of robust hydrogels
Mussels are a group of sessile aquatic bivalves from the family Mytilidae which reside in both fresh and salt waters. Saltwater mussels often live in the intertidal zone, where they are frequently bombarded with considerable stresses from waves. Mussels an-chor themselves to substrates with self-assembled fibers called byssi (singular byssus). Byssi are composed of a structure with a compliant core and a stiff cuticle, as shown in Fig. 1.1. While there are several interesting properties of mussel byssi, the key property from which we take our inspiration is the so-called “self-healing” of the byssus threads. Upon apparent plastic extensional strains of approximately 70%, the byssus appears to lose its yield point and drop in stiffness from 600 MPa to 100 MPa. However, after 1 hour of rest, the byssus thread has recovered approximately 30% of the original modulus [4, 5]. This acellular, avascular self-healing property has made byssus threads an attractive
CHAPTER 1. INTRODUCTION
area of research for the development of materials with similar characteristics.
The compliant core of byssi contains primarily collagen-like "pre-col" proteins, which have been termed "natural block copolymers" with a collagen core block flanked by two blocks rich in the amino acid histidine (His) [6]. The stiff outer shell is composed primarily of mussel foot protein-1 (mfp-1), a protein which contains a large amount of 3,4 dihydroxyphenyl-L-alanine (DOPA) [7, 8]. Both histidine and DOPA have been shown to create coordination complexes with metal ions within the mussel fibers (Fig. 1.1) [9, 10]. The coordination bonds of both histidine and DOPA have been suggested to contribute to the mechanical properties of the byssal threads by the following mech-anism: the coordinate bonds are reversible (transient) at ambient temperatures and pressures. Under stress, the coordinate bonds break sacrificially, preventing irreversible covalent bond breakage. This leads to increased toughness and strength of the mate-rial, akin to the well-known concept of “sacrificial bonds and hidden length” in biolog-ical and biomaterials fields [11]. Reformation of the coordinate bonds over time may also lead to recovery of the mechanical properties over time [4, 5, 10, 12].
Recently, researchers have taken inspiration from the mussel by using DOPA-metal or His-metal bonds as transient interactions to improve the mechanical properties of synthetic materials [12–23]. Here, we focus on Histidine:metal interactions as a model system for controlling viscoelastic material functions due to their well-studied chem-istry and lack of side reactions (as compared to DOPA).[16]
1.4
Overview of the rest of the thesis
This thesis is organized as follows: first, we present our initial findings detailing how kinetically distinct metal ions allow for a (quasi-)separation of spatial structure from temporal structure in our PEG-based hydrogels - this is the foundation of the rest of the thesis. In the following chapters, we delve into the metal-ligand chemistry and polymer physics to explore in more detail the limitations of our control over the viscoelastic
CHAPTER 1. INTRODUCTION MN NH N NH N NH CCO O C C O O C C O O M
Stiff, Hard Cuticle Compliant Core
DOPA
His Figure 1.1 | Mussels secrete groups of proteinaceous fibers called byssi to adhere to
substrates. The individual fibers, called byssal threads, are composed of a stiff, hard cuticle with a high concentration of DOPA around a more compliant core with a relatively high concentration of His. Both of these amino acids bind to transition metals, adding transient crosslinks to the byssal thread structure and contributing to the fiber’s notable mechanical properties.
Image credit: top left,Brocken Inaglory, used under aCC BY-SA 3.0 license; top right:US National Oceanic and Atmospheric Administration Great Lakes Environmental Research Laboratory.
material functions of our hydrogels. Finally, we demonstrate how the unique chemistry of Histidine:transition metal-based hydrogels can be exploited to create hydrogels with material functions which dramatically respond to longwave UV light.
In Chapter 2, we present some of the background necessary to contextualize the research presented in the following chapters. We’ll cover experimental methods for testing viscoelastic materials, metal-coordinate chemistry, and some of the theory and experimental work behind supramolecular network mechanics. We’ll also survey liter-ature from the sub-fields of metallosupramolecular network mechanics and mussel-inspired materials.
CHAPTER 1. INTRODUCTION
Chapter 3 contains the experimental methods that broadly apply to all of the chap-ters. While each chapter plays variations on the theme of controlling viscoelastic ma-terial functions, a researcher looking to either replicate or expand on the work herein should find Chapter 3 the most useful.
Chapter 4 contains our first foray into mussel-inspired material mechanics and controlling viscoelastic material functions, published as Ref. [24]. We show how kinet-ically distinct histidine:transition metal coordinate crosslinks can be used to engineer the viscoelastic properties of soft materials. This discovery lays the foundation for all of the work following.
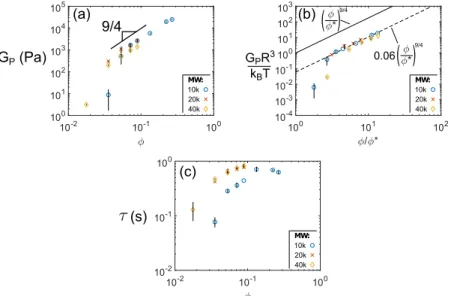
In Chapter 5, we explore how the elasticity and relaxation times of 4PEG-His:Ni hy-drogels follow from scaling arguments derived from classical semi-dilute polymer sys-tems, originally published as Ref. [25]. We further show that the moduli of these hydro-gels quantitatively follow from the equilibrium thermodynamics of the His:Ni metal-coordinate bonds. This chapter more firmly grounds the work here by comparing our results to what one might expect for the mechanical properties of our hydrogels based on the physics of conventional polymer gels.
Chapter 6 delves into the physical processes which control the relaxation timescales in 4PEG-His hydrogels by using arguments derived from Sticky Rouse theory and an experimental system wherein we use competitive ligands to coordinate the metal ions which crosslink our hydrogels.
In Chapter 7, we extend the design of our PEG-His hydrogels such that their vis-coelastic properties respond strongly to longwave UV radiation. We show that careful selection of the histidine:transition metal ion crosslink mixtures allows unique control over pre- and post-UV viscoelastic material functions, creating a family of hydrogels with programmable viscoelastic material functions.
In Appendix A, we discuss the relaxation spectrum and methods for estimatingH(τ) from experimental data. We develop a novel method based on Monte-Carlo sampling of Bayesian posterior probability distribution forH(τ). This method has the advantage
CHAPTER 1. INTRODUCTION
of assuming as little as possible about the true constitutive equation for the material and explicitly estimates the uncertainty in the estimate of the relaxation spectrum.
Appendix B is a repository of other codes used to analyze data in this thesis, includ-ing estimation of the relaxation spectrum usinclud-ing a double log-normal distribution of relaxation times, a time-temperature superposition code, code used to estimate per-colation thresholds and the gel point, and code for transposing creep moduli J(t) to the complex modulusG(ω).
1.5
Works not discussed in detail in this thesis
As a part of my graduate studies, I’ve had the pleasure to make small contributions to research that is tangentially related to my thesis research. While my contributions to these projects varied in size, I learned a great deal about soft matter chemistry, physics, and biophysics through these experiences, and discussions with my co-authors below heavily influenced my thoughts about my thesis work. Publications based on these works can be found in:
1. K. Kawamoto, S. C. Grindy, J. Liu, N. Holten-Andersen, and J. A. Johnson. “Dual Role for 1,2,4,5-Tetrazines in Polymer Networks: Combining DielsâĂŞAlder Re-actions and Metal Coordination To Generate Functional Supramolecular Gels”.
ACS Macro Letters 4.4 (2015), pp. 458-461.
2. P. Chen, Q. Li, S. C. Grindy, and N. Holten-Andersen. “White-Light-Emitting Lan-thanide Metallogels with Tunable Luminescence and Reversible Stimuli-Responsive Properties”. Journal of the American Chemical Society 137.36 (2015), pp. 11590-11593.
3. D. Mozhdehi, J. A. Neal, S. C. Grindy, Y. Cordeau, S. Ayala, N. Holten-Andersen, and Z. Guan. “Tuning Dynamic Mechanical Response in Metallopolymer Net-works through Simultaneous Control of Structural and Temporal Properties of
CHAPTER 1. INTRODUCTION
Chapter 2
Background
2.1
Time-Dependent Mechanical Properties:
Viscoelas-ticity and Rheology
When measuring the mechanical performance of an engineering material, by far and away the most useful test is the tensile test, so pervasive that it’s taught to freshman engineering students of every stripe. By measuring the characteristic stress-strain re-sponse of the material, we determine the material’s elastic (or Young’s) modulusE and the strength σy. In effect, the Young’s modulus describes the linear load-deformation relationship for the material, and the strength tells us to what extent that linear rela-tionship is valid. These measurements are typically (implicitly) assumed to be mea-sured during a quasi-static process, and there is no time-dependence to their values. In practice the time-dependence of these materials properties is very small.
Conversely, the mechanical properties of many soft materials are strongly time-dependent, and therefore the conventional tensile test is not sufficient to describe their mechanical properties in the linear regime. Indeed, the deformation and flow prop-erties of materials can be subdivided into further categories with more exotic behav-ior than simple linear-elastic (Hookean) solids or linear-viscous (Newtonian) fluids.
CHAPTER 2. BACKGROUND Solid Fluid Twilight Zone Twilight Zone
Plasto-Elastic Elastic
Visco-Inelastic Newtonian Bingham Plasto-Inelastic Non-Bingh am Recoverable Hookean Non-Hookean Recoverable Ideal
Non-Ideal Non-Ideal
Non-New tonian Incom pletely Recov erab le Incom pletely Recov erable Flow Elastic Deformation Plastic Viscous
Figure 2.1 | A reproduction of the original rheological chart originally from Bilmes [26]. The chart is divided into fluid-like (upper half ) and solid-like materials (lower half ), with a “Twilight Zone” of not-quite fluid and not-quite solid materials to the left and right.
In 1942, the British Rheologists’ Club published “A Rheological Chart” in Nature[26], categorizing the variously observed types of behavior. A reproduction of this chart is shown in Figure 2.1. Clearly, boring, linear behavior only constitutes a small fraction of the types of mechanical behavior which we can expect.
As the mechanical properties of a given materials class become less ideal and move towards the Twilight Zone, solving a materials design problem using that material be-comes increasingly difficult. This is due to the fact that interpreting the microscopic-to-molecular scale significance of each type of abnormal deformation becomes com-mensurately difficult. In this thesis, we focus on one Twilight Zone class of materials: viscoelastic materials. We have identified some strategies for designing materials with
CHAPTER 2. BACKGROUND
specific viscoelastic properties using transient metal-coordinate bonds as introduced in Chapter 1 and as detailed below. While viscoelastic materials represent only one sub-set of complex mechanical behaviors, it is our hope that our work inspires researchers to undertake more detailed studies on the physical mechanisms behind the various complex types of deformation in order to exploit their complexity to design functional materials.
2.1.1
Mechanical Tests for Viscoelastic Materials
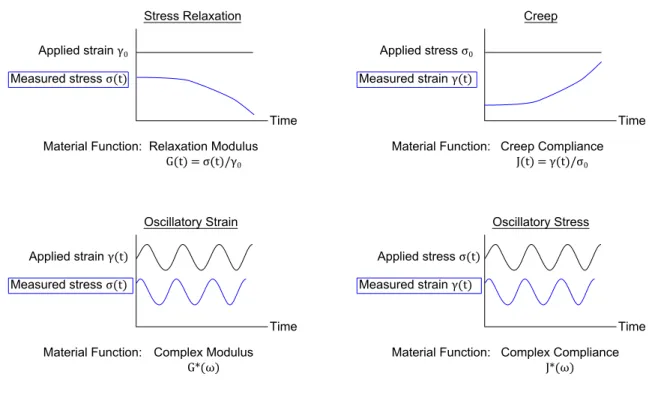
Since the conventional test for engineering materials, the tensile test, will not suffice to properly describe the mechanical properties of viscoelastic materials, we need to use more complicated techniques to measure the time-dependent viscoelastic mate-rial functions of our soft matemate-rials. Four of these test are outlined in Figure 2.2: Stress Relaxation, Creep, Oscillatory Strain, and Oscillatory Stress. For soft materials, these measurements are typically performed in a shear geometry using an instrument called a rheometer.
Of these four, the most common are Stress Relaxation, Creep, and Oscillatory Strain. By convention of the field, oscillatory stress tests are simply not commonly performed. In a stress relaxation experiment, we apply a constant strain γ = γ0and measure the
stress σ(t) as a function of time. The material function we effectively measure during this experiment is the relaxation modulusG(t), defined as σ(t)/γ0. In an analogous
manner, during a creep experiment we apply a constant stress σ = σ0, and we
mea-sure the strain γ(t) as a function of time. Oscillatory tests are more complicated: we apply a sinusoidal strain γ(t) = γ0sin(ωt) with angular frequency ω, and in the linear
regime we measure a sinusoidal stress that has an identical frequency ω but may be phase-shifted by an amount δ: σ(t) = σ0sin(ωt+δ). We convert this measurement into
the complex modulusG∗by converting the sinusoidal stress into two components: one that is in-phase with the applied strain and one that is out-of-phase with the applied strain. We then define the two parts of the complex modulus to describe the
magni-CHAPTER 2. BACKGROUND
Applied strain γ0
Stress Relaxation
Measured stress σ(t)
Time Material Function: Relaxation Modulus
G(t) = σ(t)/γ0
Applied stress σ0
Creep
Measured strain γ(t)
Time Material Function: Creep Compliance
J(t) = γ(t)/σ0
Applied strain γ(t)
Oscillatory Strain
Measured stress σ(t)
Time Material Function: Complex Modulus
G*(ω)
Applied stress σ(t)
Oscillatory Stress
Measured strain γ(t)
Time Material Function: Complex Compliance
J*(ω)
Figure 2.2 | The four common mechanical tests for viscoelastic materials. Here, we define shear strain as γ and shear stress as σ.
tude of the in-phase and out-of-phase components: G0for the in-phase part (called the storage modulus) andG00for the out-of-phase part (called the loss modulus), such thatG∗ =G0+iG00.
σ(t) = σ0sin(ωt + δ) (2.1)
σ(t) = σ0{sin(ωt)cos(δ) + cos(ωt)sin(δ)} (2.2) G0≡ σ0 γ0cos(δ); G 00 ≡ σ0 γ0sin(δ) (2.3) σ(t) = γ0{G 0 sin(ωt) +G00cos(ωt)} (2.4)
And by measuringG0andG00at various frequencies, we measure the material func-tionG∗(ω). Each of the four tests shown in Figure 2.2 have unique advantages and disadvantages in terms of sensitivity, straightforwardness of data interpretation, and experimental duration. However, in the linear (low-strain) regime, each of these
ex-CHAPTER 2. BACKGROUND
periments provide equivalent information. For example, in principle one can uniquely convert a measurement of the relaxation modulusG(t) to the complex modulusG∗(ω) and vice versa. In this thesis, most of the experimental data will be presented as oscil-latory strain measurements because they are the most transparently feature-rich when it comes to analyzing soft hydrogels (as we will demonstrate below). However, oscilla-tory strain experiments can be extremely time consuming when measuring data at low frequencies. For example, at a frequency of ω = 10−3rad/s, one period of oscillation lasts T ≈ ω−1 = 103 s. Typically, the rheometer needs ∼10 periods of oscillation to confidently measure the modulus. Therefore, just to measure the modulus at ω = 10−3 rad/s, it will take 10T = 104s ≈ 3 hours! Since we want to measure the material func-tionG∗at many frequencies, this is not appropriate if we are concerned with (e.g.) sam-ple dehydration. However, a creep experiment that measures J(t) at timest1 <t < t2
s can be converted into the complex modulusG∗(ω) over the intervalt2−1 < ω < t1−1. Therefore, if we perform a creep experiment at times up tot = 103s, we can convert our measuredJ(t) intoG∗(ω) with a minimum frequency of ωmi n ≈ 10−3rad/s. In effect, we are trading any inaccuracy caused by the long duration of our oscillatory measurement for some inaccuracy caused by the (often numerical) transformation of J(t) →G∗(ω), and we pick up the advantage of increased experimental efficiency. Much of the data in this thesis is from a combination of oscillatory measurements at high frequencies and transformed creep measurements at low frequencies (long times).
2.1.2
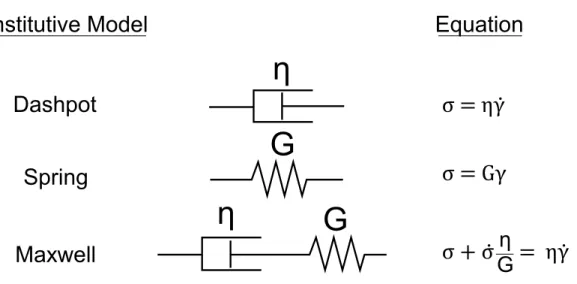
Phenomenological Models for Viscoelastic Materials
When examining experimental data from mechanical experiments on viscoelastic ma-terials, we often try to reduce the dimensionality of the material functions by fitting them to a constitutive model. Constitutive models are, put simply, relationships be-tween the stress and strain of a material, as well as the time derivatives of the stress or strain. For example, for Hookean solid materials the constitutive model is a linear proportionality between the stress and strain like a spring; the proportionality
con-CHAPTER 2. BACKGROUND
η
G
η
G
Constitutive Model
Dashpot
Spring
Maxwell
Equation
σ = ηγ
σ = Gγ
σ + σ = ηγ
η
G
Figure 2.3 | Constitutive models allow us to quantitatively estimate materials parameters for comparison between materials and possibly extrapolate from the
measured data.
stant is the elastic modulus, i.e. σ = Gγ. For a Newtonian fluid, the relationship is a proportionality between the stress and strain-rate: σ = η ˙γ; a mechanical analogue for a Newtonian fluid is a dashpot. Constitutive models can then be fit to experimental data to quantify a material’s behavior, providing a facile means of comparison between materials.
For viscoelastic materials, we often use combinations of springs and dashpots to represent the solid-like and fluid-like nature of viscoelastic materials. The geometry of how we combine them dictates the final form of the constitutive relationship. The simplest of these, with a spring and dashpot in series, is called the Maxwell model. Its constitutive relation is shown in Figure 2.3.
While Figure 2.3 shows only one spring in series with one dashpot, in principle any number of springs and dashpots in any arrangement may be used as a constitutive model for a viscoelastic material. However, any arrangement of these simple elements is equivalent to the “canonical form” of springs and dashpots [27, 28], which can be rep-resented as a parallel array of an arbitrary number of Maxwell elements seen in Figure 2.4.
CHAPTER 2. BACKGROUND
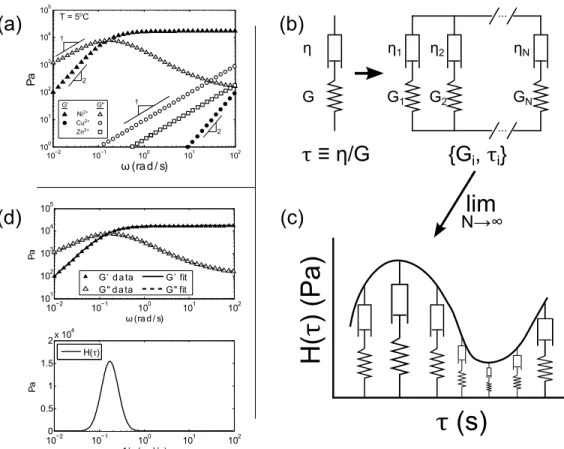
τ (s)
H(
τ)
(P
a)
N→∞lim
η G{G
i, τ
i}
τ ≡ η/G
η1 G1 ηN G2 GN ... ... η2Figure 2.4 | Any combination of springs and dashpots is equivalent to the “canonical model,” shown at center. Taking the limit of an infinite number of springs and dashpots, the canonical model becomes the relaxation spectrum as described in the text.
In oscillatory measurements, the Maxwell model predicts the following (with the relaxation time τ defined as τ ≡ η/G):
G0(ω) = Gω 2τ2
1 + ω2τ2 (2.5)
G00(ω) = Gωτ
1 + ω2τ2 (2.6)
as shown graphically in Figure 2.5. The canonical model is then simply a sum over the parallel Maxwell elements:
G0(ω) = X i Giω2τi2 1 + ω2τi2 (2.7) G00(ω) = X i Giωτ 1 + ω2τi2 (2.8)
In the special case where an infinite number of parallel Maxwell elements are used as the constitutive model, the discrete set of relaxation times and moduli {τi,Gi} (each corresponding to a single Maxwell element) becomes a continuous distribution of
re-CHAPTER 2. BACKGROUND
laxation modes, and we define the relaxation spectrumH(τ):
G0(ω) = Z ∞ −∞ H(τ) ω 2τ2 1 + ω2τ2dlnτ (2.9) G00(ω) = Z ∞ −∞ H(τ) ωτ 1 + ω2τ2dlnτ (2.10) More discussion of the relaxation spectrum and why it may or may not be an actually useful material function to compute is available in Chapter 4 and Appendix A.
The features of the Maxwell model are clearly visible from a measurement of the storage and loss moduli when presented on a log-log scale, as in Figure 2.5, left. We can observe τ as the inverse of the frequency whereG0=G00, or equivalently ω−1whereG00 is at a local maximum. This maximum in energy dissipation allows us to think of τ as the inverse of the resonant frequency of the system. The plateau modulusG can be seen as the high-frequency limit of the storage modulus. Besides the relaxation time and plateau modulus, another feature is the scaling of the moduli in the regime ω << τ−1, called the terminal regime. There, we expect the relationshipsG0
(ω) ∝ ω2and G00(ω) ∝ ω1. This can be seen by taking the limit of Equations 2.5 as ω → 0.
Even when our material is represented by multiple Maxwell elements (as in the canonical model), the relaxation times, plateau moduli, and terminal scaling behavior are still visible from oscillatory data, as shown in Figure 2.5. This relative ease in identi-fying viscoelastic mechanical signatures is what makes oscillatory deformation exper-iments the most useful method for relating material structure to mechanical function.
2.2
Supramolecular Polymeric Hydrogels
Heretofore, the information contained in this chapter is material agnostic - it can ap-ply regardless of the microstructure of the material. As introduced in Chapter 1, in this thesis we specifically study hydrogels composed of polymer chains which interact with each other primarily through transient, reversible interactions. This subclass of soft
CHAPTER 2. BACKGROUND 10-310-210-1100101102103 Angular Frequency 10-3 10-2 10-1 100 101 102 Modulus / P a 10-310-210-1100101102103 Angular Frequency 10-3 10-2 10-1 100 101 102 10-310-210-1100101102103 Angular Frequency 10-3 10-2 10-1 100 101 102 1 2 1 2 G' G'' τ = 1 s G = 1 Pa τ-1 G τ1 = 1 s, G1 = 1 Pa τ2 = 50 s, G2 = 10 Pa τ-1 τ-1 2 1 τ1 = 1 s, G1 = 1 Pa τ2 = 0.05 s, G2 = 10 Pa τ-1 τ-1 1 2 (c) (b) (a)
Figure 2.5 | (a) Under oscillatory deformation, the Maxwell model shows that the two parameters are directly identifiable from experimental data (without necessarily fitting the constitutive model): (1) The relaxation time τ,
observable as ω−1whereG0=G00, or equivalently ω−1at the local maximum inG00; (2) The plateau modulusG, observable as the high-frequency limit of
G0. At frequencies ω << τ−1, the moduli are expected to follow the
relationshipsG0∝ω2andG00∝ ω1.
(b,c) In models with multiple relaxation modes, the observables from each Maxwell element take on slightly different forms depending on the relative relaxation times and moduli for each mode, but nonetheless a frequency sweep experiment clearly identifies the signatures of each relaxation mode.
materials is called “supramolecular,” since the structure is defined by intermolecular interactions. In this section, we review some of the canonical theory of supramolecular relaxation as well as experimental studies on the mechanical properties of supramol-ecular polymeric materials, both in the gel state (swollen with solvent) and the melt state (in the absence of solvent).
Transient interactions can vary widely in bond strength and stoichiometry; the tran-sient bonds can be very weak such as hydrogen bonds or with strengths that approach the strength of covalent bonds. Examples of transient bonds used to improve the me-chanical properties of soft materials include hydrogen bonds, ionic bonds [29, 30], hydrophobic interactions [31, 32], adaptable covalent bonds, protein associating do-mains and other bio-inspired associations [33–36], host-guest interactions [37], and metal-coordinate bonds [18, 24, 25, 38–47]. Each of these motifs vary widely in bind-ing energy, kinetics, stoichiometry, stimuli-responsiveness, ease of chemical synthesis,
CHAPTER 2. BACKGROUND
and cost of precursors, but they share the property of introducing energy-dissipative properties into a polymer network. In particular, supramolecular materials have found promising use as a component of tough hydrogels, where the goal is maximizing the strength, toughness, and durability of a material made up largely by water. One promi-nent example of these materials is double-network hydrogels, originally discovered by Gong et al. [48]. One strategy for designing tough double-network hydrogels is to em-bed a supramolecular network inside a loosely- but permanently-crosslinked network. Many types of supramolecular chemistries have been used to engineer tough, double-network hydrogels; a review of many of these strategies is available in Zhao [3].
One of the goals of the field of supramolecular network materials is to identify the set of unifying physical principles that apply to every type of supramolecular transient associations. Despite the advances in the study of the use of transient interactions to engineer soft material properties, there is still much to be learned about the fundamen-tal physical principles which govern the limits of materials design through transient interactions, especially in the context of bulk mechanical properties.
2.2.1
The Theory of Transient Networks
In 1946, Green & Tobolsky published what I believe is one of the first examples of a molecular theory for polymeric viscoelasticity, entitled “A New Approach to the Theory of Relaxing Polymeric Media”[49]. Therein, they showed that a polymeric material with a single bond-breaking rate should have the constitutive equation identical to that of the Maxwell model, and the parameter that sets the relaxation time is the inverse of the bond dissociation rate constantkd, i.e. τ ≈kd−1. Green and Tobolsky assumed that the bond breaking and reforming occurred at the same rate; this constraint was relaxed by Tanaka and Edwards [50] for un-entangled polymers.
In 2001, Rubinstein and Semenov [51] published a comprehensive overview of the “Sticky Rouse” and “Sticky Reptation” theories, which account for reversibly binding interactions across polymer concentration regimes. They give scaling predictions for
CHAPTER 2. BACKGROUND
the relaxation time across a wide array of polymer concentrations with respect to poly-mer size, spacing between reversibly associating “stickers,” and polypoly-mer concentra-tion.
In the context of this thesis, an important concept that they propose is the renor-malization of the reversible bond lifetime, described as follows: the key process that relaxes stress within a supramolecular material is when a supramolecular interaction dissociates and each binding partner finds a new complementary functional group to bind to. In this way, the polymeric network relaxes stress by rearranging the network topology into a stress-free state. The rate of this process (effectively the material’s re-laxation time) is controlled by the supramolecular dissociation kinetics, in line with Green and Tobolsky’s original formulation. However, when a supramolecular func-tional group separates from its partner, it may not find a new partner to bind to if the concentration of open groups is low enough. Therefore, the open group will simply re-bind to its original partner and be unable to effectively relax any stress from the net-work. Depending on the concentration of open functional groups, the effective bond lifetime may be 10-100 or more times longer than the intrinsic bond lifetime. We will use this idea in Chapter 6 to understand the relaxation processes in the hydrogels we discuss here.
2.2.2
Kinetically-Controlled Supramolecular Materials
Two seminal experimental works in the field of supramolecular viscoelastic materials are those of Annable et al. [52] and Yount, Loveless, and Craig [38]. In Annable et al., they studied hydrogels composed of telechelic block copolymer systems with the ge-ometry A-B-A, where A represents a hydrophobic block and B represents a hydrophyllic block. At sufficiently high polymer concentrations, the hydrophobic blocks form mi-celles, which crosslink the solutions into hydrogels. The A blocks can dissociate from a micelle and join a neighboring micelle, effectively acting as transient crosslinks. What Annable et al. found is that the length of the hydrophobic block directly correlated
CHAPTER 2. BACKGROUND
to the macroscopic mechanical relaxation time of the hydrogel: longer hydrophobic blocks led to materials with longer relaxation times. Additionally, they found that by making hydrogels with multiple distinct hydrophobic block lengths, they could create materials with multiple relaxation times, and that by controlling the relative concen-tration of each end-block length they were able to control the modulus associated with the corresponding relaxation time.
Yount, Loveless, and Craig used a different materials system to arrive at a similar conclusion. They used poly(4-vinyl pyridine) crosslinked with either platinum or pal-ladium “pincer” complexes, and found that by sterically hindering the pincer com-plex near the metal center, they were able to alter the pyridine-metal exchange ki-netics without significantly altering the thermodynamics of the process. This gave them a set of complexes with relatively similar binding energies (as determined by the equilibrium constants) and orders of magnitude differences in dissociation constants. Through this materials system, they were able to experimentally confirm that the dis-sociation rate constant effectively controlled the relaxation time of the material. In a later study [42], they were able to show that by combining pincer complexes with dis-tinct kinetics, they were able to tune the relaxation modes in a manner analogous to Annable et al.
2.3
Metal-coordinate chemistry
2.3.1
Metal-Coordination Thermodynamics and Kinetics
Metal-coordination complexes are molecules formed when one or more atoms donate a nonbonding pair of electrons to a metal ion center, forming a coordinate covalent
bond. The electronic structure of these complexes tend to have strong absorption
bands in the visible range, and therefore coordination complexes have brilliant, varied colors. Molecules whose atoms donate the electrons to the metal ion to form the co-ordinate bond are called ligands. Metal-coco-ordinate complexes are sometimes referred
CHAPTER 2. BACKGROUND
to using the shorthand MxHyLz, where M represents the metal and H represents the
protonation state of the ligand L.
The strength of metal-coordinate bonds can vary greatly depending on the ligand and metal center, from very weak bonds (logK ≈ 0 − 1) to very strong, effectively permanent bonds (logK ≈ 40), whereK is the equilibrium constant for the reaction L + M −−−→ ML. However, across many ligand species, the trend of complex stability almost always follows the order of the Irving-Williams series [53]: Mn < Fe < Co < Ni < Cu > Zn. Indeed, for a variety of ligands-metal combinations pertinent to this thesis, this trend nearly always applies as seen in Figure 2.6.
Co2+
Cu2+
Ni2+
ML MHL
Figure 2.6 | Equilibrium constants for a series of ligands and metals, showing the Irving-Williams series relationship. For the pyrophosphate ligand, we separated the data into those from ML complexes and MHL complexes as identified in the figure. All data is from the NIST Standard Reference Database No. 46 [54].
CHAPTER 2. BACKGROUND
Intriguingly, the kinetics of metal-coordinate complexes also have a consistent trend across ligands: for a given ligand, the trend of kinetic rate constants follows the same order of metals, much like the Irving-Williams series dictates the order of complex sta-bility for metal ions. However, this series does not follow the same order as the Irving-Williams series. For example, across the first-row transition metals, the rate constant
kH2O for the ligand exchange reaction M(H2O)n + H2O* −−−→ M(H2O)n-1H2O* + H2O follows the trend Ni < Co < Fe (II) < Mn < Cr (II) < Cu [55], which clearly does not follow the trend of the Irving-Williams series. For the transition metals studied in this thesis, the trend holds across a wide variety of ligands as shown in Figure 2.7.
k
1 − 1Figure 2.7 | Ligand exchange rate constants for a series of ligands and metals, showing the trendkC u >kC o >kN i (contrast this with Figure 2.6, which says that
KC u >KN i > KC o). All data is from Helm and Merbach [56]. Abbreviations
are en: 1,2 diaminomethane, MeOH: methanol, tn: 1,3 propanediamine, MeCN: acetonitrile, DMF: N,N’-dimethylformamide, DMSO: dimethyl sulfoxide, AcOH: acetic acid, meea: 2-methoxyethylamine, TMU: N,N,N’,N’-tetramethylurea, pa: n-propylamine.
CHAPTER 2. BACKGROUND
coworkers [38, 42] showed that using transient interactions with kinetically distinct timescales allows one to engineer the viscoelastic properties of supramolecular poly-mer gels. Materials which employ metal-coordination as the supramolecular motif can change the thermodynamics and kinetic properties of their systems by several orders of magnitude simply by swapping out which metal ion binds to the coordinating ligands. Thus, in the spirit of the work done by Annable et al. and Stephen Craig and coworkers, in this thesis we’ll endeavor to create supramolecular hydrogels where the viscoelastic mechanical properties can be controlled in a facile manner by switching out one metal ion for another.
2.4
Metallosupramolecular Network Mechanics
In this section, we review recent advances in using metal-coordination to control the mechanical properties of supramolecular networks, a sub-field called “metallo-supramolecular networks.” A significant amount of the development in this field came from the group of Stuart J. Rowan, primarily studying polymers conjugated to 2,6-bis(N-methylbenzimidazolyl) pyridine) (MeBip). MeBip strongly binds to a variety of metal ions in a tridentate fashion with two imidazolyl and one pyridyl nitrogens all forming coordinate bonds with a single metal center. MeBip tends to form com-plexes with transition metals withML2 stoichiometry and lanthanide ions with ML3 stoichiometry, and Rowan and coworkers used these complexes to crosslink polymer systems [57, 58]. In addition, the MeBip:M complexes can phase-separate from the rub-bery polymer domains, leaving a two-phase morphology in these systems. By mixing a transition metal (Zn2+) with a lanthanide (Eu3+), they were able to control the extent of chain extension vs. crosslinking and therefore the resultant mechanical properties of the networks [59]. In particular, using more Zn2+and less Eu3+causes the relaxation time of the network to decrease substantially.
CHAPTER 2. BACKGROUND
by Burnworth et al. [60]. They used UV-A irradiation of ca. 1000 mW/cm2to heal defects in the material. The MeBip:M complexes absorb quite strongly in the UV-A regime, and the absorbed energy is then converted to heat. The heat locally raises the temperature in the proximity of the MeBip:M crosslinks, accelerating their exchange dynamics and allowing scratches and cracks to be repaired. Further functionality was incorporated into MeBip:M networks by Miller et al. [61]. In MeBip:Cu crosslinked networks, they showed that oxidizing Cu1+ −−−→ Cu2+ using nitroso tetrafluoroborate as an oxidant could decrease the specific viscosity by approximately 50%. Using ascorbic acid to re-duce Cu2+ −−−→ Cu1+returned the specific viscosity to its original value.
Another well-known family of metallosupramolecular networks is based on terpyri-dine:metal crosslinks, much of which have been studied in the group of Sebastian Seif-fert. For example, they showed that using Mn2+, Zn2+, or Co2+ could alter the mate-rial’s relaxation times in accordance with established trends on metal-ligand exchange kinetics [45]. They also showed that the choice of solvent can significantly alter the network dynamics. The polymer diffusivity and dynamic light scattering relaxation showed similar trends with respect to metal ions [44].
Other groups have found ways to modify the oxidation states of metal centers in metallosupramolecular materials in order to control the mechanical properties. For example, Sato et al. [62] showed that, in a poly(ethylene glycol)-based hydrogel cross-linked with phosphate-Fe2+coordinate bonds, adding the oxidant H2O2induced a sol-to-gel transition due to the oxidation of Fe2+ −−−→ Fe3+. Addition of ascorbic acid to reduce Fe3+ −−−→ Fe2+reversed the change in mechanical properties. Giammanco and Ostrowski [63] exploited the chemistry of uronate-Fe coordination to use ca. 400-500 nm light to reduce Fe3+−−−→ Fe2+, modifying the network structure and decreasing the shear modulus by ∼50%.
Besides chemical oxidizing agents and photochemical techniques, electrostatic po-tentials can also be used to change the oxidation state of metal ions. Palleau et al. [64] applied a voltage across a sodium polyacrylate-based hydrogel using a copper wire as
CHAPTER 2. BACKGROUND
the cathode. At the cathode, copper was oxidized from Cu0 −−−→ Cu2+and dissolved into the hydrogel. Cu2+forms coordinate bonds with the carboxylic acid groups of the sodium polyacrylate, adding coordinate crosslinks to the hydrogel in the vicinity of the copper wire. Palleau et al. calls this process ionoprinting, since the wire is used to locally print ions into the hydrogel.
2.5
Metal-Coordination in Biology and Bio-inspired
Ma-terials: The Mussel Byssus Thread
2.5.1
Metal-Coordination in the byssus
As we introduced in Chapter 1, the mussel byssal thread employs metal-coordinate bonds to enhance the mechanical properties of its threads, originally discovered by Vaccaro and Waite [4]. Here, we summarize more recent investigations of the role of metal-coordination in mussel threads.
The mussel thread is primarily composed of proteins which self-assemble during the creation of the thread. In this process, the mussel sticks its appendage called a “foot” down to the target substrate. Then, glands inject precursor proteins into the ventral groove of the foot. The proteins then self-assemble into the plaque, which ad-heres to the substrate, and thread, which connects the plaque to the mussel’s tissue. Detailed reviews of the processes and proteins that form both the adhesive plaque and thread are available in Lee et al. [22] and Waite [65].
As discussed in Chapter 1, the byssal thread has a core-shell structure, with a stiff, hard outer coating called the cuticle covering a more compliant core [66]. The cuti-cle itself has a two-phase morphology composed of granules embedded in a matrix. The cuticle is primarily composed of mussel foot protein 1 (“mfp1”), a protein with ap-proximately 15mol% 3,4 dihydroxyphenyl-L-alanine (DOPA)[22], an amino acid that is the result of a post-translational modification of tyrosine. DOPA can form coordinate
CHAPTER 2. BACKGROUND
bonds with a wide variety of metal ions. In the two-phase structure of the cuticle, the DOPA tends to be found in the granules, where it forms coordinate bonds that harden the cuticle, which has been shown to contribute to the excellent mechanical perfor-mance of the byssal threads [10].
Besides its ability to form metal-coordinate bonds, DOPA has a very high affinity for surfaces, and current thinking among biologists is that DOPA plays a significant role in the strong adhesive properties of the plaque. For example, Lee, Scherer, and Messersmith [67] found via atomic force microscopy that the breaking strength of a single DOPA adhering to a Ti surface is ∼800 pN, roughly half the strength of a covalent bond. However, the DOPA-surface interaction was fully reversible and repeatable [67].
Finally, in addition to metal-coordinate bonds, DOPA can undergo a wide variety of chemical reactions depending on the pH and presence of nucleophiles. One in-triguing example of DOPA’s functionality is polydopamine, a polymer made from DOPA monomers. Polydopamine has been shown to adhere to surfaces in a very robust man-ner, and is relatively facile to synthesize and control [68].
The proteins in the thread core are very different than those found in the cuticle and in the plaque. The bulk of the core is composed of collagen-like proteins called “pre-Cols.” Pre-Cols are composed of 3 basic regions: (1) a center block that has a collagen structure (with amino acid repeats of the form [Gly-AA2-AA3], where AAn represents
any amino acid. This block self-assembles into triple-helix structures much like typical collagens. The collagen block is flanked by a block whose properties change depending on the type of pre-Col, but some contain elastin-like amino acid sequences X-Gly-X-Pro-Gly, where X = Gly or a nonpolar amino acid. Finally, the flanking domains are further surrounded by a histidine-rich domain (HRD), with up to ∼20mol% His (Figure 2.8) [22]. In the HRD, histidines form coordinate bonds with transition metals, further reinforcing the mechanical properties of the mussel byssus [14, 69–71].
CHAPTER 2. BACKGROUND Collagen-like domain Flanking domain Flanking domain Histidine-rich domain Histidine-rich domain M N NH N NH N NH
His
Figure 2.8 | The structure of Pre-Col proteins which compose the thread core. Histidine-rich domains (HRDs) contain ∼20mol% His, which forms coordinate complexes with transition metals.
2.5.2
Mussel-Inspired Metal-Coordination in Synthetic Materials
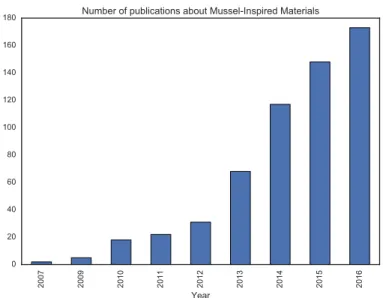
The uniquely excellent mechanical properties of the mussel byssal thread and plaque have recently lead to a flood of researchers making synthetic materials inspired by the chemistry and structure of the mussel byssus. Since 2007 there has been a large increase in the number of peer-reviewed publications on the topic of mussel-inspired materials (Figure 2.9). The focus of many of these papers is on exploiting the multi-functionality of DOPA to create materials with strong adhesive and cohesive properties, and primar-ily focused on biomaterials as the application. In fact, DOPA-based tissue sealants have compared favorably to industry-standard adhesives in both sealing performance and cytotoxicity [72]. A much smaller body of work has focused on Histidine-based materi-als. In this section, we review a selection of the literature on mussel-inspired synthetic materials that incorporate metal coordination.
CHAPTER 2. BACKGROUND
Figure 2.9 | In recent years, there has been a large increase in the research area of
mussel-inspired materials. Data obtained on March 25, 2017 from a search in Thomson Reuters Web of Science for the query: TOPIC: (MUSSEL-INSPIRED) OR TOPIC: ("MUSSEL INSPIRED") OR TOPIC: (("BIO INSPIRED" OR
"BIO-INSPIRED") AND "MUSSEL").
DOPA-Metal Interactions in Synthetic Materials
The first mussel-inspired material to employ metal coordination was by Holten-Andersen et al. [12], where a four-arm poly(ethylene glycol)-DOPA conjugated polymer was shown to make hydrogels at physiologic pH when mixed with Fe3+. The relaxation time was shown to depend strongly on the pH, with higher pH leading to longer relaxation times. This was attributed to shifting the equilibria of the DOPA-Fe3+ complexes, where at high pH (>10), a 3:1 DOPA:Fe3+complex is the preferred state, while at medium pH (6-8.5) a 2:1 complex is preferred, and at low pH a 1:1 complex is preferred. These mate-rials were later extended to use DOPA:Al3+and DOPA:V3+, and the relaxation time was shown to also depend strongly on the choice of metal ion [21]. Menyo, Hawker, and Waite [73] showed that the catechol moiety of DOPA could be modified by adding elec-trophilic groups on the benzyl ring while maintaining the bidentate phenolate nature of the catechol, and the resulting hydrogels were able to have mechanical rigidity at lower pH due to the reduced pKa of the hydroxyl groups and be more stable to
oxida-CHAPTER 2. BACKGROUND
tion.
In addition to four-arm poly(ethylene glycol), DOPA-metal coordinate bonds have been incorporated into a variety of soft material architectures, including peptide nanofibers [13], chitosan [19, 74], poly(hydroxyethyl acrylamide) [75], gelatin [76], and using metal oxide nanoparticles instead of metal ions as crosslinks [18]. A notable advance in ma-terial synthesis made by Lee and coworkers is to ionoprint Fe3+ions on the surface of a hydrogel by applying a voltage across a DOPA-containing hydrogel using an Fe wire as the cathode; this process allows local control over the DOPA:Fe crosslink concentration [75, 77].
His-Metal Interactions in Synthetic Materials
Fullenkamp et al. [23] was the first study to make hydrogels crosslinked by His:metal bonds, using the four-arm poly(ethylene glycol) conjugated to an N-terminal histidine. Fullenkamp et al. showed that (1) the primary amine group participates in the complex: hydrogels did not form when using deamino-histidine as the crosslink; (2) by using dif-ferent metal ions as the crosslinker, the hydrogels’ relaxation times shifted in a pattern corresponding to the expected ligand exchange rates, in concordance with the infor-mation presented above in Figure 2.7, i.e. τN i > τC o > τC u > τZ n.
Since that work, His:metal interactions have also been added to other architec-tures, including a polystyrene-polybutylacrylate thermoplastic elastomer [78], resilin (an elastomeric biopolymer) [79], telechelic polymers with both His:metal bonds and hydrophobic associations [43], and isolated mussel protebased films [80]. An in-triguing extension of the functionality of these His:metal-based materials was made by Wegner et al. [81], who showed that His:Co2+ crosslinks could be oxidized to His:Co3+ crosslinks using H2O2, dramatically altering the viscoelastic properties due to the in-ertness of Co3+-based complexes.
![Figure 2.1 | A reproduction of the original rheological chart originally from Bilmes [26].](https://thumb-eu.123doks.com/thumbv2/123doknet/14418936.513185/30.918.216.707.165.623/figure-reproduction-original-rheological-chart-originally-bilmes.webp)