Chaperoning viral protein evolution
by
Angela Marie Phillips
B.S. in Chemistry (2012)
University of Florida, Gainesville, FL
Submitted to the Department of Chemistry
in Partial Fulfillment of the Requirements for the Degree of
Doctor of Philosophy in Biological Chemistry
at the
Massachusetts Institute of Technology
June 2018
@
2018 Massachusetts Institute of Technology
All rights reserved
Signature of Author:
Signature redacted
Department
d
Chemistry
MY
7,20
1 8Certified by:
Signature redacted
Matthew D. Shoulders
Whitehead Career Development Associate Professor
Thesis supervisor
Accepted by:
Signature redacted
Robert W. Field
Chairman, Department Committee on Graduate Students
This doctoral thesis has been examined by a committee of the Department of Chemistry as
follows:
Signature redacted
\K)
John M. Essigmann
William R. (1956) & Be
sy
P. Leitch Professor in Residence
Professor of Chemistry, Toxicology, and Biological Engineering
Director, MIT Center for Environmental Health Sciences (CEHS)
Department of Chemistry, and Department of Biological Engineering, MIT
Thesis committee chair
_00 lo-1 0_
Signature redacted
Matthew D. Shoulders
Whitehead Career Development Associate Professor
Associate Member, Broad Institute at Harvard and MIT
Investigator, Center for Skeletal Research at Massachusetts General Hospital
Member, MIT Center for Environmental Health Sciences
Department of Chemistry, MIT
Thesis supervisor
Signature redacted
0'
Leonid A. Mirny
Professor of Medical Engineering and Science, and Physics
Associate Member, Dana-Farber Cancer Institute
Associate Member, Broad Institute at Harvard and MIT
Institute of Medical Engineering and Science, and Department of Physics, MIT
Thesis committee member
Chaperoning viral protein evolution
by
Angela Marie Phillips
Submitted to the Department of Chemistry on May 7, 2018 in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Biological Chemistry
ABSTRACT
Preventing viral pandemics and developing effective antiviral therapeutics demands understanding the molecular mechanisms that both potentiate and constrain viral evolution. The rapid evolution of viruses is mediated in part by their high mutation rates, enabling resistance to antiviral drugs, seasonal vaccines, and innate and adaptive immune responses. Fortunately for us, the same mutations responsible for resistance are often biophysically deleterious to viral proteins. Thus, viral evolution is inherently constrained by the proper folding of viral proteins into functional, stable conformations. In cells, protein folding and homeostasis are assisted by complex networks of chaperones and quality control machinery. Though the evolutionary implications of most chaperones and quality control factors remain unexplored, the HSP90 chaperone can buffer and potentiate the phenotypic effects of mutations in endogenous client proteins in bacteria, fungi, plants, and other eukaryotic organisms. Viruses acquire mutations at a rate several orders of magnitude above that of the aforementioned organisms, yet they do not encode any machinery to assist destabilized protein variants to their folded, functional conformations. However, viral proteins are known to interact with host chaperones and quality control machinery. My graduate work has focused on determining whether and how host proteostasis machinery modulates viral protein evolution. First, I employed a serial passaging approach to evolve influenza in host cells with remodeled proteostasis capacities, revealing that cytosolic host proteostasis capacity is indeed a critical determinant of influenza evolutionary trajectories. This work motivated systematic quantification of influenza protein mutational tolerance upon perturbation of host proteostasis, for which I applied deep mutational scanning
to comprehensively profile the mutational tolerance of influenza nucleoprotein and
hemagglutinin in modulated cytosolic and endoplasmic reticulum (ER) folding environments, respectively. The nucleoprotein work provides the first experimental evidence that host chaperones can enhance the accessibility of biophysically deleterious, adaptive viral protein variants. The hemagglutinin work establishes evolutionary implications for the ER proteostasis machinery, and demonstrates that ER proteostasis mechanisms enhance mutational tolerance across the entire HA protein. Overall, it is clear that host chaperones and quality control
Chapter Abstracts
Chapter 1. Protein homeostasis and evolution at the host-pathogen interface
The evolution of host and viral proteins is mediated by missense mutations that can endow new function, which are often biophysically deleterious. Thus, evolution is necessarily constrained by protein stability and folding. In host cells, protein-folding challenges are addressed by proteostasis networks composed of chaperones and quality control factors that work in concert to shepherd nascent proteins to folded, functional conformations. Work focused primarily on the HSP90 chaperone has suggested a critical role for chaperones in modulating the evolution of their endogenous clients, in part by buffering biophysically deleterious effects of non-synonymous mutations. Viral genomes, which acquire mutations at a rate several orders of magnitude above that of both prokaryotes and eukaryotes, typically do not encode endogenous chaperones or other co-factors to assist protein folding. However, viral proteins do engage host chaperones, and host chaperone inhibitors have been shown to limit the viability of certain RNA viruses. While there is substantial evidence that viral protein evolution is constrained by stability, the possibility that host chaperones can shape the evolution of viral pathogens had not been studied before this thesis. Here, we review progress towards understanding the impact of endogenous proteostasis machinery on client protein evolution, and furthermore, what consequences this machinery has on the evolution of invading pathogens.
Chapter 2. Host proteostasis modulates influenza evolution
Predicting and constraining RNA virus evolution require understanding the molecular factors that define the mutational landscape accessible to these pathogens. RNA viruses typically have high mutation rates, resulting in frequent production of protein variants with compromised biophysical properties. Their evolution is necessarily constrained by the consequent challenge
to protein folding and function. We hypothesize that host proteostasis mechanisms may be significant determinants of the fitness of viral protein variants, serving as a critical force shaping viral evolution. Here, we test this hypothesis by propagating influenza in host cells displaying chemically-controlled, divergent proteostasis environments. We find that both the nature of selection on the influenza genome and the accessibility of specific mutational trajectories are significantly impacted by host proteostasis. These findings provide new insights into features of host-pathogen interactions that shape viral evolution, and into the potential design of host proteostasis-targeted antiviral therapeutics that are refractory to resistance.
Chapter 3. Host chaperones buffer the fitness of destabilized adaptive influenza variants The threat of viral pandemics demands a comprehensive understanding of evolution at the host-pathogen interface. Here, we systematically show that the accessibility of adaptive mutations in influenza nucleoprotein at fever-like temperatures is mediated by host chaperones. Particularly noteworthy, we observe that the Pro283 nucleoprotein variant, which is conserved across human influenza strains, confers resistance to the MxA antiviral-restriction factor, and played a key role in the adaptation to humans of the 1918 pandemic influenza strain, is rendered unfit by host chaperone depletion. This fitness loss is linked to biophysical defects that chaperones cannot address when heat shock factor-I is inhibited. Thus, host chaperones can uncouple biophysically deleterious consequences of mutations from the benefits of immune escape. In summary, host chaperones play a central role in shaping influenza adaptation, with implications for the evolution of other viruses, for viral host-switching, and for the design of
Chapter 4. ER proteostasis and temperature modulate the mutational tolerance of influenza hemagglutinin
Influenza virus hemagglutinin (HA) evolves rapidly to escape antibodies, but also performs an essential role in viral replication. Therefore, the virus's rapid evolution is contingent on HA possessing sufficient mutational tolerance to acquire antibody resistance while continuing to properly fold and assemble in the ER of host cells. Here, we investigate the mutational tolerance of HA in host cells with modulated ER protein homeostasis (proteostasis) machinery, at normal body temperature and at a biophysically challenging, fever-like temperature. We find that upregulation of ER proteostasis machinery generally enhances HA mutational tolerance, and that increased temperature generally reduces HA mutational tolerance. Intriguingly, variants that are most temperature-sensitive, which are likely also biophysically problematic, benefit most from upregulation of proteostasis machinery. Overall, this work demonstrates that host ER proteostasis mechanisms and temperature modulate HA mutational tolerance, and reports the first evidence of evolutionary implications for the ER proteostasis machinery.
Chapter 5. Conclusions and future directions
The findings reported in this thesis are briefly summarized, and possible future directions for investigating the evolutionary implications of maintaining proteostasis are discussed.
Some days the positive path is harder to find and we have to be relentless in its pursuit. But a better outlook is always there and worth chasing. On the other side are potential-and
possibility.
Acknowledgements
The work presented in this thesis would not have been remotely possible without many people. First and foremost, I need to thank my advisor, Prof. Matthew Shoulders. Throughout my Ph.D., Matt has led by example, pushing me to become a more creative, efficient, and cognizant scientist. I think Matt would agree that most of the techniques and expertise required for my
Ph.D. research were not in our lab's wheelhouse five years ago. Daily, Matt made sure I had everything I needed to move forward, whether that meant obtaining materials from another lab, taking a course, or even being co-advised. His support was essential for my progress, and ultimately enabled us to answer fundamental questions about protein evolution. Beyond the daily experimental grind, I am so thankful that Matt involved me in his grant writing, which was one of my favorite parts of graduate school, constantly provided me with opportunities to present my work, and allowed me to mentor Luna, Anna, and Apolonia. Matt also encouraged me to pursue my interests outside the lab-such as advocacy trips to D.C. and running the
12 2nd Boston Marathon-for which I am greatly appreciative. Finally, I need to thank Matt for
sharing his perspective on pursuing an academic career and for providing critical feedback on my fellowship and post-doctoral application packages. I know that I'll continue to look to Matt for advice throughout my career, and hope that I can become such an effective scientist and mentor.
Next, I need to thank some of my other mentors who have supported my scientific career. Thanks to Prof. Leonid Mirny, for welcoming me into his lab, for providing essential feedback on my research, and for supporting my post-doctoral search. Thanks to Prof. John Essigmann, for preparing me for my qualifiying exam, for inspiring me to be a better teacher, and for always making me step back and think more broadly. I must also thank Leonid and John for taking the time to be on my thesis committee-they both have provided meaningful feedback throughout my Ph.D., and for that I am very thankful. I'd also like to thank my undergraduate advisors-Profs. Laura Bohn and William Ja-who continued to support me throughout graduate school, writing many letters for graduate programs and fellowship applications, and offering their advice whenever I needed it.
Throughout graduate school I have had the privilege to participate in many productive collaborations. I'd like to thank Jesse Bloom and the Bloom Lab, especially Orr Ashenberg and Mike Doud, for welcoming me into their lab, for helping me implement an approach that transformed my Ph.D. research, and for providing feedback and reagents faster than I could ask for them. I am also indebted to Vincent Butty and Stuart Levine at the MIT BioMicro Center, for helping us design our sequencing projects, analyzing massive amounts of sequencing data to meet grant deadlines, and troubleshooting sequencing anomalies. I am also grateful for our collaboration with Yu-Shan Lin's Lab at Tufts, especially Sean McHugh and Jiayuan Miao, who did the molecular dynamics simulations in Chapters 2 and 3. Special thanks to Yu-Shan for her tireless efforts to compile a pipeline for the data analysis presented in Chapter 4-her energy is inspiring. I also need to thank Alex Shalek and Jon Trombetta for introducing us to library prep and sequencing, and for their patience and connections to the necessary resources.
welcoming me into the viral evolution project and helping me hit the ground running; Luna, for the ingenuity and honest skepticism she brought to our project; Anna, for her energy and motivation in pushing our project forward; Louis and Chris R., for chasing me down Beacon street into my last mile of the Boston Marathon; and my neighbor of four years-Chet, for his enthusiastic criticism and active listening skills.
Thanks to my colleagues on the Science Policy Initiative Exec. team-for teaching me so much over the past two years, for the time they invest in SPI, and for the support from the MIT administration and MIT D.C. office that make our work possible.
Thanks to my fellow WIC Admin members for making WIC the professional development organization it is today, and for the relationships WIC fosters between women in the Chemistry Department.
Next, I need to thank my friends, for making graduate school a great time. Alex, Wade, and Justin, thanks for dragging me out for 9pm champagne, for weekends of board games, biking, and road trips, and for not leaving Boston (at least for now). Drennan Lab, thanks for adopting me, for margs at Border and pitchers at Newtown, and for greatly improving my interview talk. Cullen, Sydney, and Jenny, thanks for sending a care package when I got my first F, and for always providing sound advice. Joanna, Chloe, Kiplyn, and Reagan, thanks for your support and confidence, and for your inspiring strength and impact.
Thanks to my family, not just for supporting me remotely, but for encouraging me throughout my education and for having unyielding confidence in my abilities. Thanks to my Mom, for instilling a fierce determination in me that has powered me through the many long days that make up a Ph.D. Thanks to my Dad, for his ruthless commitment to routine that has ingrained the time management skills essential for my productivity. Thanks to Julie, for her energy and selflessness that has kept us so close despite being so far apart. Thanks to Hailey, for her inspirational strength and grit. Thanks to Joe, for those one-liners that always come at the right time. Thanks to Matt, for always having a solution when something was broken. Special thanks to Mom, Dad, and Julie, for the daily phone calls on my walk home and for visiting Boston so many times (Hailey and Joe, my post-doc at Harvard buys you more time). Thanks to my Aunt Jenny and Uncle Rich, for providing a relaxing getaway from MIT and the city.
Last but certainly not least, I need to thank Kenny Kang, for supporting me everyday, scientifically and personally. Over the past five years, Kenny has listened to dozens of practice talks, helped me work through numerous confusing results, and has never hesitated to give his honest advice. Though he rarely cared to rant about his day, he always let me unload my daily stress on him. All of the success I've had has been supported by his love, confidence, and selflessness. I must also thank Kenny's family for being so welcoming, for entertaining several ambitious vacation itineraries, and for old vine zinfandels.
I'd finally like to thank Matt, Madeline, Cullen, Andrew, Chris R., Kenny, Luna, and Chet, for feedback on the chapters in this thesis, and the National Science Foundation for funding the last three years of my Ph.D.
Table of Contents Abstract 3 Chapter Abstracts 4 Acknowledgements 9 Table of Contents 11 List of Tables 14 List of Figures 15 List of Abbreviations 18
Chapter 1: Protein homeostasis and evolution at the host-pathogen interface
1.1 Summary 27
1.2 The host-pathogen interface 28
1.3 Protein evolution at the host-pathogen interface 29
1.4 Host proteostasis mechanisms assist host protein folding 32
1.5 Host proteostasis both constrains and potentiates host protein evolution 35
1.6 Viruses engage host proteostasis factors 38
1.7 Host proteostasis mechanisms potentiate and constrain viral protein evolution 40
1.8 Summary and future directions 42
1.9 References 45
Chapter 2: Host proteostasis mechanisms modulate influenza
2.1 Author Contributions 51
2.2 Introduction 52
2.3 Results 54
Small molecule-based strategies create three distinctive host proteostasis 54 environments for influenza evolution experiments
Serial passaging to emulate influenza evolution 61
Nature of selection pressure differs in modified proteostasis environments 63
Influenza protein mutational landscapes are modulated by host proteostasis 71
qPCR 81
RNA-Seq 82
RNA-Seq analysis 82
Cellular thermal shift assay (CETSA) 83
Serial passaging and hemagglutination-based titering 84
Infectious viral titering via tissue culture infectious dose (TCID50) assay 85
Reverse genetics 85
Deep sequencing 85
Sequencing data analysis 86
Statistics 87
Molecular dynamics simulations 88
2.6 Acknowledgements 91
2.7 References 92
Chapter 3: Host chaperones can buffer the fitness of destabilized adaptive influenza variants
3.1 Author Contributions 98
3.2 Introduction 99
3.3 Results 101
Deep mutational scanning of nucleoprotein in a biophysically challenging 101
environment
Elucidating the cause of compromised fitness of WT residues in a biophysically 113
challenging environment
Balancing protein biophysical properties with immune escape 117
3.4 Discussion 119
3.5 Materials and Methods 122
Plasmids 122
Antibodies 122
Cell lines 122
Cell line characterization 123
Deep mutational scanning 125
Pairwise viral competitions 127
Molecular dynamics simulations 128
Biophysical characterization 130
Data availability 131
Statistics 131
3.6 Acknowledgements 134
Chapter 4: ER proteostasis and temperature modulate influenza hemagglutinin mutational tolerance
4.1 Author Contributions 139
4.2 Introduction 140
4.3 Results 143
Modulating ER proteostasis during influenza infection 143
Evaluating the impact of ER proteostasis mechanisms on a known trafficking- 150
defective HA variant
Deep mutational scanning of HA in modulated ER proteostasis environments 152
4.4 Discussion 167
4.5 Materials and Methods 169
Plasmids 169
Cell culture 169
Influenza virus 169
Compounds and antibodies 169
qPCR 170
RNA-seq 170
Deep mutational scanning 172
Deep mutational scanning data analysis 173
Infectious viral titering via tissue culture infectious dose (TCID50) assay 173
Reverse genetics viral competitions 174
Pairwise competition sequencing data analysis 175
Flow cytometry 175
Statistics 176
4.6 Acknowledgements 179
4.7 References 180
Chapter 5: Conclusions and future directions
5.1 Conclusions 184
5.2 Future directions 187
5.3 References 191
List of Tables
Chapter 2: Host proteostasis mechanisms modulate influenza evolution
Table 2. 3. 1 List of ten highest % frequency non-synonymous mutations for each proteostasis en vironm ent and p assage...68 Table 2. 3. 2 List of ten highest % frequency synonymous mutations for each proteostasis en vironm ent and p assage...69 Table 2. 3. 3 Molecular Dynamics Simulations: Energy Contributions...74 Table 2. 5. 1 Primer sequences for qPCR and PA sequencing ... 81
Chapter 3: Host chaperones can buffer the fitness of destabilized adaptive influenza variants
Table 3. 5. 1 List of primers for qPCR and sequencing library preparation. ... 133
Chapter 4: ER proteostasis and temperature modulate influenza hemagglutinin mutational tolerance
List of Figures
Chapter 1: Protein homeostasis and evolution at the host-pathogen interface
Figure 1. 3. 1 Biophysical boundary model of protein evolution ... ... ... 30
Figure 1. 4. 1 Protein folding energy landscape... ... 32
Figure 1. 4. 2 Eukaryotic chaperone-assisted protein folding... ... 33
Figure 1. 5. 1 Genetically diverse populations respond to inhibition of chaperone buffers (A) and
potentiators (B) ... . _ .... 35
Figure 1. 5. 2 Chaperones can shift the boundary of accessible protein stabilities. ... ... 37
Figure 1. 6. 1 Influenza proteins interact extensively with host proteostasis factors... 38 Figure 1. 7. 1 Model for impact of chaperones on viral protein evolution.. ... ... 41 Chapter 2: Host proteostasis mechanisms modulate influenza evolution
Figure 2. 3. 1 Chemical biology methods to modify the host cell's proteostasis environment ... 56
Figure 2. 3. 2 Validation of chemical biology tools used to perturb proteostasis ... 57
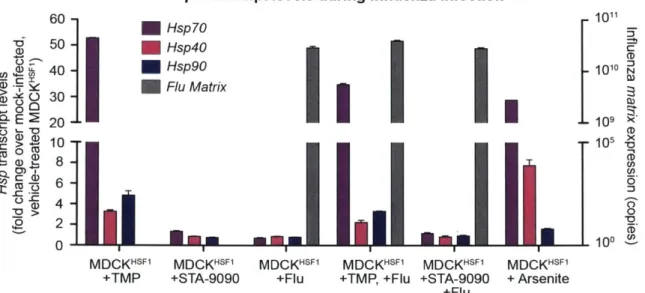
Figure 2. 3. 3 Heat shock protein transcript expression during influenza infection in modulated
p ro te o sta sis e n viro n m e n ts ... ... ... ... ... 5 8
Figure 2. 3. 4 Transcriptomic analysis of perturbed host cell proteostasis environments ... 60 Figure 2. 3. 5 Serial passaging of Influenza A /Wuhan/95 H3N2 ... ... 61
Figure 2. 3. 6 Multiplicity of infection and hemagglutination titers during serial passaging ... 62
Figure 2. 3. 7 Site frequency spectra show frequency distribution of non-synonymous (A) and synonymous (B) mutations in a given folding environment at a particular passage...66 Figure 2. 3. 8 Trajectories for non-synonymous (A) and synonymous (B) mutations that increase in frequency during serial passaging ... ... 6 7
Figure 2. 3. 9 Analysis of non-synonymous mutations observed in distinctive proteostasis environments. Aligned variants were observed in any of three biological replicates ... 72
Figure 2. 3. 10 HA and PA display divergent mutational trajectories in HSF1-activated versus basal versus H sp90-inhibited environm ents ... 76
Chapter 3: Host chaperones can buffer the fitness of destabilized adaptive influenza variants
Figure 3. 3. 1 Transcriptional profiles of modulated host environments... ... 102 Figure 3. 3. 2 HSFIi is effective during influenza infection and does not significantly perturb influenza propagation or host cell metabolic activity ... 103
Figure 3. 3. 3 Deep mutational scanning reveals positively selected sites upon chaperone
depletion at a restrictive tem perature ... ... 104 Figure 3. 3. 4 Pairwise competition recapitulates deep mutational scanning batch competition Figure 3. 3. 5 Full sequence logo plot for nucleoprotein HSF1-inhibited environment at 39 'C
relative to a basal environment at 37 'C . . 107
Figure 3. 3. 6 Full sequence logo plot for nucleoprotein: HSF1-inhibited environment at 39 'C
relative to a basal environment at 39 C .... . 108
Figure 3. 3. 7 Full sequence logo plot for nucleoprotein: 39 'C relative to 37 'C in a basal
e n viro n m e n t ... ... ... .. 10 9
Figure 3. 3. 8 Full sequence logo plot for nucleoprotein: HSF1-inhibited environment at 37 'C
relative to a basal environment at 37 'C ... 110
Figure 3. 3. 9 Full sequence logo plot for nucleoprotein: Hsp90-inhibited environment at 39 'C
relative to a basal environment at 39 'C . ... 111
Figure 3. 3. 10 Pairwise competitions recapitulate deep mutational scanning batch
com p etition ... ... 1...112
Figure 3. 3. 11 Pro283 disrupts nucleoprotein a-helical content and is destabilized relative to variants at site 283
... 1 15 Figure 3. 3. 12 Purification and thermal denaturation of recombinant nucleoprotein variants ...116
Figure 3. 3. 13 Host chaperones modulate immune escape of Pro283 nucleoprotein...117 Figure 3. 3. 14 Host chaperones mediate the accessibility of biophysically destabilized adaptive m u ta tio n s ... ... ... 1 1 8
Chapter 4: ER proteostasis and temperature modulate influenza hemagglutinin
mutational tolerance
Figure 4. 2. 1 The unfolded protein response is a stress-responsive integrated signaling
Figure 4. 3. 1 Induction of XBP1s and A TF6f/XBPIs is selective and does not cause global
stress...
... ... ... 145Figure 4. 3. 2 Full transcriptome analysis confirmed selectivity of XBP1s and A TF6f/XBP1s
induction ... ... 147
Figure 4. 3. 3 Methods to induce XBPIs and A TF6f/XBPIs are functional during influenza
infection ... .. ... ... 149
Figure 4. 3. 4 YI 74H-HA viral growth and surface expression are affected by XBPIs
induction...
...
. ... 152Figure 4. 3. 5 Deep mutational scanning of HA in modulated ER proteostasis environments ...153
Figure 4. 3. 6 ER proteostasis mechanisms and temperature impact HA global mutational
to le ra n c e ... ... . .. ... .. ... . . ... ... 15 5
Figure 4. 3. 7 HA variants are depleted upon increased temperature and enriched upon XBPIs
and A TF6f/XBPIs induction. . ... ... .. 156
Figure 4. 3. 8 Divergent fitness effects revealed by DMS were validated by pairwise
competitions... . 158
Figure 4. 3. 9 Selection on HA imposed by XBPIs induction is opposite that of increased
temperature.. . . 159
Figure 4. 3. 10 Mutational tolerance at sites across HA is impacted by XBPIs induction and
increased temperature ... . 161
Figure 4. 3. 11 Differential selection on WSN HA in a basal environment at 39 'C relative to 37
'C was quantified by DMS... - . . 162
Figure 4. 3. 12 Differential selection on WSN HA upon XBPIs induction at 37 'C was quantified
by D M S ... ... ... 163
Figure 4. 3. 13 Differential selection on WSN HA upon XBPIs induction at 39 'C was quantified
by D M S .. ... ... 16 4
Figure 4. 3. 14 Differential selection on WSN HA upon A TF6f/XBPls induction at 37 'C was
quantified by DMS . .. . 165
Figure 4. 3. 15 Differential selection on WSN HA upon A TF6f/XBPls induction at 39 'C was
List of Abbreviations 0C AGfolding
A
A
A280
ACTA2adp
AMDHD2 ARL4CATF4
ATF6
ATF6f
ATP
BAG2 BAG3 BAG5BH
BHLHA 15 BiPBSA
C
CALR CAMK I CANX CCR7CD
CHACIcHSF1
CHOP
cDNA
CDS
Degrees Celsius
Energy of folding
Angstrom
Alanine
Absorbance at 280 nm
Actin
Adjusted p-value
Amidohydrolase domain containing 2
ADP-ribosylation factor-like 4C
Activating transcription factor 4
Activating transcription factor 6
Transcriptionally-active activating transcription factor 6
Adenosine triphosphate
BLC2 associated anathogene 2
BLC2 associated anathogene 3
BLC2 associated anathogene 5
Benjamin-Hochberg-adjusted p-value
Basic helix-loop-helix family member Al 5
Binding immunoglobulin protein
Bovine serum albumin
Cysteine
Calreticulin
Calcium/calmodulin dependent protein kinase I
Calnexin
C-C motif chemokine receptor 7
Circular dichroism
ChaC glutathione specific gamma-glutamylcyclotransferase 1
Constituitive heat shock factor 1
CCAAT-enhancer-binding protein homologous protein
Complementary deoxynucleic acid
CETSA Cellular thermal shift assay
cHSF1 Constitutive heat shock factor 1
CLDNI Claudin 1
CLU Clusterin
CMV Cytomegalovirus
COPI Coat protein 11
COX3 Cytochrome C oxidase subunit 3
CRELD2 Cysteine rich with EGF (epidermal like growth factor) like domains 1 CYPIBI Cytochrome P450 family 1 subfamily B member 1
D Aspartate
DAPI 4',6-diamidino-2-phenylindole
DDIT4 DNA damage inducible transcript 4
DERL3 Degradation in endoplasmic reticulum protein 3
DHFR Diydrofolate reductase
diffsel Differential selection
DMEM Dulbecco's modified eagle medium
DMS Deep mutational scanning
DMSO Dimethyl sulfoxide
DNA Deoxynucleic acid
DNAJA 1 DnaJ heat shock protein family (Hsp40) member Al
DNA JA2 DnaJ heat shock protein family (Hsp40) member A2
DNAJA3 DnaJ heat shock protein family (Hsp40) member A3
DNAJBI DnaJ heat shock protein family (Hsp40) member B1
DNAJB2 DnaJ heat shock protein family (Hsp40) member B2
DNAJB6 DnaJ heat shock protein family (Hsp40) member B6
DNA JB8 DnaJ heat shock protein family (Hsp40) member B8
DNAJB9 DnaJ heat shock protein family (Hsp40) member B9
DNAJB 11 DnaJ heat shock protein family (Hsp40) member B1 1
EDEMI EDTA EEF1A EELE elF2a EIF2AK3 EIF5 EINT ER ERAD Erg3 ERQ1B EVDW f F FAM19A4 FBS FBXW1O FGF21 Fl6v3 F/CD FKBP14 FKBP4 FPLC fwd G GADD34 GAPDH GBSA GEO GJA I GRP94 GSG GSOL-POLGB
ER degradation enhancing alpha-mannosidase like protein 1 Ethylenediaminetetraacetic acid
Eukaryotic translation elongation factor 1 alpha 1 Coulombic energy
Eukaryotic translation initiation factor 2A
Eukaryotic translation initiation factor 2 alpha kinase 3 Eukaryotic translation initiation factor-5
Internal energy
Endoplasmic reticulum
Endoplasmic reticulum-associated degradation S. cerevisiae C-5 sterol desaturase
Endoplasmic reticulum oxidoreductase 1 beta Van der Waals energy
Mutation frequency Phenylalanine
Family with sequence similarity 19 member A4 Fetal bovine serum
F-box and WD repeat domain-containing 10 Fibroblast growth factor 21
F domain broadly neutralizing HA antibody
FIC domain containing (Huntingtin-interacting protein) FK506 binding protein 14
FK506 binding protein 4
Fast protein liquid chromatography Forward
Glycine
Growth arrest and DNA damage-inducible protein Glyceraldehyde-3-phosphate dehydrogenase Generalized Born and surface area
Gene Expression Omnibus Gap junction protein alpha 1 Glucose-regulated protein 94 kDa Glycine-serine-glycine
GSOL,NP
H
h
H1
HA
HEK
HERPUD I HIDIHIV
H1N1
H3N2
hpi
HSF1
HSP
HSP40
HSP70
HSP90
HSP90AA I HSP90AA2 HSPA2 HSPA4 HSPA4L HSPA5 HSPA6 HSPA1A HSPAIB HSPAIL HSPA8 HSPA9Nonpolar solvation free energy Histidine
Hour(s)
Hemagglutinin subtype 1 influenza Hemagglutinin
Human embryonic kidney
Homocysteine inducible ER protein with ubiquitin like domain 1
High-temperature-induced Dauer formation domain containing Human immunodeficiency virus
Hemagglutinin subtype 1, neuraminidase subtype 1 influenza Hemagglutinin subtype 3, neuraminidase subtype 2 influenza Hours post-infection
Heat shock factor 1 Heat shock protein Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock Heat shock protein protein protein protein protein protein protein protein protein protein protein protein protein protein 40 70 90 90 90 kDa kDa kDa
alpha family class A member 1 alpha family class A member 2 family A (Hsp70) member 2
family
family
family
family
family
family family familyHeat shock protein family
(Hsp70) member 4 (Hsp70) member 4 like (Hsp70) member 5 (Hsp70) member 6 (Hsp70) member 1A (Hsp70) member 1 B (Hsp70) member 1 like (Hsp70) member 8 (Hsp70) member 9
IFIT1 IgG IPTG IRE1 IRES ISG15 ISG20 K kcal/mol KDEL KDELR3 KRT27 LB M M MANIBI MD MDCK MDCK-SIAT1 MIA mL MMP13 MOI mRNA MUC20 mut MWCO MxA MX1 MX2 N NA NEP NP
Interferon induced protein with tetratricopeptide repeats 1 Immunoglobulin G
Isopropyl P-D-1-thiogalactopyranoside
Inositol-requiring enzyme 1 Internal ribosomal entry site
Interferon stimulated exonuclease gene 15 Interferon stimulated exonuclease gene 20 Lysine
Kilocalories per mole
Lysine-aspartate-glutamate-leucine
KDEL endoplasmic reticulum protein retention receptor 3 Keratin 27
Lysogeny broth Matrix
Methionine
Mannosidase alpha class 1B member 1 Molecular dynamics
Madin Darby canine kidney cells
Madin Darby canine kidney cells expressing a-2,6-sialic acid receptors Melanoma inhibitory activity
Milliliters
Matrix metallopeptidase 13 Multiplicity of infection Messenger ribonucleic acid Mucin 20
Mutant
Molecular weight cut off Myxovirus resistance protein 1 MX dynamin like GTPase 1 MX dynamin like GTPase 2 Asparagine
Neuraminidase Nuclear export protein Nucleoprotein
NQOI
NAD(P)H quinone dehydrogenase 1
ns
Not significant
NS1
Non-structural protein 1
OAS2
2'-5'-Oligoadenylate synthetase 2
OD
Optical density
P
Proline
PA
Polymerase acidic unit
PARP3
Poly(ADP-ribose) polymerase family member 3
PBS
Phosphate buffered saline
PBSA
Phosphate buffered saline with BSA
PB1
Polymerase basic unit 1
PB2
Polymerase basic unit 2
PCDH7
Protocadherin 7
PCR
Polymerase chain reaction
PDB
Protein database
PDI
Protein disulfide isomerase
PDIA1
Protein disulfide isomerase family A member 1
PD/A3
Protein disulfide isomerase family A member 3
PD/A6
Protein disulfide isomerase family A member 6
PDL
Poly-D-lysine
pDZ
Post synaptic density protein domain
PERK
Protein kinase RNA-like endoplasmic reticulum kinase
pH
Potential of hydrogen
PIN4
Peptidylprolyl cis/trans isomerase
PLPP5
Phospholipid Phosphatase 5
pme
Posterior mean estimates
PME
Particle mesh Ewald
PR8
Puerto Rico 8
RBC
RBM3rev
RI
RIPA
RNA
RNase H
RNA-seq
RPKM
RPL36A RPLP2RSA
S
Sip
S2P
S. cerevisiae
SDF2L I
SDS
SDS-PAGE
SE
SEC24D
SEC6IAl
SEM
SERPI
SerpinH1
SFS
SIAT1
SLC
SNP
SPINK5
SPP1
SS
ssRNA
Pearson correlation coefficient Red blood cell
RNA-binding motif protein 3 Reverse
Recombinant inbred
Radioimmunoprecipitation assay buffer Ribonucleic acid
Ribonuclease H
Ribonucleic acid sequencing Reads per kilobase million Ribosomal protein L36a
Ribosomal protein lateral stalk subunit P2 Relative surface accessibility
Serine
Site 1 protease Site 2 protease
Saccharomyces cerevisiae
Stromal cell derived factor 2 Like 1 Sodium dodecyl sulfate
Sodium dodecyl sulfate polyacrylamide gel electrophoresis Site entropy
SEC24 homolog D, COPlI coat complex component Sec6l translocon alpha 1 subunit
Standard error of the mean
Stress associated endoplasmic reticulum protein 1 Serpin family H member 1
Site frequency spectrum
a-2,6-Linked sialic acid receptor-expressing Solute carrier family member
Single nucleotide polymorphism
Serine peptidase inhibitor, kazal type 5 Secreted phosphoprotein 1
Secondary structure
STC Staniocalcin 1
STR Short tandem repeat
syn Synonymous
T Threonine
TCID50 Tissue culture infectious dose 50
Tm Apparent melting temperature
ts Temperature sensitive
TMP Trimethoprim
TPCK L-1-tosylamide-2-phenylethyl chloromethyl ketone
TRIB3 Tribbles Pseudokinase 3
TRPA1 Transient receptor potential ankyrin 1
UCSC University of California, Santa Cruz
UGGT1 UDP-glucose glycoprotein glucosyltransferase 1
UPR Unfolded protein response
W Tryptophan
WSN Influenza virus A/WSN/1 933
WT Wild-type
XBP1 X-box like protein 1
XBP1s Spliced X-box like protein 1
Y Tyrosine
Chapter 1: Protein homeostasis and evolution
at the host-pathogen interface
1.1
Summary
The evolution of host and viral proteins is mediated by missense mutations that can endow new function, which are often biophysically deleterious. Thus, evolution is necessarily constrained by protein stability and folding. In host cells, protein-folding challenges are addressed by proteostasis networks composed of chaperones and quality control factors that work in concert to shepherd nascent proteins to folded, functional conformations 1-3 Work focused primarily on the HSP90 chaperone has suggested a critical role for chaperones in modulating the evolution of their endogenous clients 4-1, in part by buffering biophysically deleterious effects of non-synonymous mutations. Viral genomes, which acquire mutations at a rate several orders of magnitude above that of both prokaryotes and eukaryotes, typically do not encode endogenous chaperones or other co-factors to assist protein folding. Instead, viral proteins engage host chaperones to assist the folding of their proteins 14-18, and host chaperone inhibitors have been
shown to limit the viability of certain RNA viruses 1923. While there is substantial evidence that viral protein evolution is constrained by stability, the possibility that host chaperones can shape the evolution of viral pathogens had not been studied before this thesis 24-27. Here, we review
progress towards understanding the impact of endogenous proteostasis machinery on client protein evolution, and furthermore, what consequences this machinery has on the evolution of invading pathogens.
1.2 The host-pathogen interface
Hosts have evolved diverse signaling pathways to sense and respond to viral infections. For instance, the innate immune system fights viral infection by killing infected cells, degrading viral genomic material, and inhibiting viral replication and translation 2-30. In response, viruses have
evolved mechanisms to evade these host systems, including mutating epitope regions to evade host recognition and activating inhibitors of the innate immune response 29.30 If the host and
virus both survive initial infection, the virus may be cleared by the adaptive immune response, which elicits virus-specific neutralizing antibodies. Alternatively, the viral infection may persist if the virus acquires mutations that enable it to escape neutralization 31. Thus, the host and virus
are continuously co-evolving. The host must eliminate the invading pathogen while maintaining organismal viability. In contrast, viruses are minimalist pathogens that rely on their host's viability to propagate. Therefore, viruses must strike a delicate balance of hijacking host machinery and evading the host immune response without inducing widespread cytotoxicity.
1.3 Protein evolution at the host-pathogen interface
The evolution of host and viral proteins is in part mediated by missense mutations that can enhance protein fitness. For example, amino acid substitutions can enable viral escape from neutralizing antibodies, or increase the antiviral activity of host restriction factors. Often, these missense mutations diminish protein thermodynamic and kinetic stability and, hence, activity and organismal fitness.
Protein evolvability is therefore constrained by stability. Most globular proteins are marginally stable, with a AGfolding of approximately -10 kcal/mol 32,33. The high dimensionality of
protein sequence space clusters viable sequences in this marginally stable regime. Very few sequences have higher stability, while numerous sequences have lower stability 34. It follows that amino acid substitutions are often destabilizing 35. Moreover, random walk lattice models, in concert with biochemical reconstitution, have demonstrated that stable proteins tend to be more evolvable, presumably because they can accommodate more mutations and still maintain their functional folds 36,37. Experimentally, a combination of biochemical and biophysical tools has 37-40
identified protein stability as a molecular signature of protein fitness and protein function
-Phylogenetic analyses validated by ancestral protein reconstruction have established that the thermodynamic stability of viral and host protein variants corresponds to their inferred fitness based on evolutionary sequence data 3738,40
Moreover, protein folding has been implicated as the mediator between protein stability and protein evolvability. Proteins must fold in a reasonable amount of time into a functional form that is stable to side-reactions like aggregation and proteolysis 34. Random walk lattice models of protein evolution have revealed that protein evolutionary trajectories are confined to neutral
can be phenotypically silent at other positions if they do not impact protein folding or function
.
Theoretical work has attributed the covariation between sequence evolution, codon usage, and
mRNA levels across many organisms to selection against misfolded proteins
45.Experimental
work on adenylate kinase demonstrated that protein fitness is not solely dependent on enzyme
activity, but also on resistance to denaturation and aggregation
46.Additionally, divergent RNase
H sequences have conserved folding pathways that are more kinetically stable than their most
recent common ancestor
47.These studies, among many others, support a model in which the
evolutionary accessibility of protein variants depends not only on inherent thermodynamic
stability, but also on kinetic stability, defined by folding and misfolding rates (Figure 1. 3. 1).
This biophysical constraint has significant implications for viruses, which acquire
mutations at rates several orders of magnitude above that of eukaryotes
48.Such high mutation
rates enable viruses to rapidly explore amino acid space as they adapt to new environmental
pressures,
but are often
detrimental to
viral
fast
folding rate
propagation. Viral genomes are small, largely protein
slow
coding, and frequently contain overlapping reading
fast
%
eframes.
Thus, the deleterious effects of a single
misfolding
rate
mutation are often amplified in a viral genome
slow
These deleterious effects can be relieved, however, by
stability
stabilizing compensatory mutations
5
or by assistance
stable
Figure 1. 3. 1 Biophysical boundary from more fit viral genomes 49,51.
model of protein evolution.
Because protein evolution is constrained by
The accessibility of protein variants is
dictated by thermodynamic and kinetic
stability, destabilizing adaptive variants often must be
(folding
versus
misfolding
rate)
stability. Protein variants inside the
preceded or accompanied by permissive mutations
biophysical boundary are accessible,
and those outside the boundary, are
that are thermodynamically or kinetically stabilizing
.
inaccessible. Adapted from Powers et
a/.3
Frequently, these stabilizing mutations are neutral in
the absence of the destabilizing adaptive mutation, as selection demands proteins meet the
stability threshold, but does not favor increased stability above that threshold. Thus, permissive stabilizing mutations occur stochastically, making specific protein evolutionary trajectories unpredictable 5.53. Still, numerous cases of host and viral proteins exhibiting this idiosyncratic
epistasis, when the effects of one mutation are influenced by another, have experimentally demonstrated that protein thermodynamic stability and the kinetic stability of folding intermediates are critical constraints of protein evolution 38,40,50,54 This is particularly evident for
influenza proteins. For example, influenza nucleoprotein was not able to tolerate destabilizing immune escape mutations until stabilizing permissive mutations occurred 4 , and influenza neuraminidase was unable to tolerate a drug resistance mutation unless it was accompanied by a mutation that rescues neuraminidase surface expression 39. Thus, epistasis is pervasive in protein evolution, and is often explained by protein stability or folding and assembly kinetics.
Another important consideration in examining the constraints on protein evolution is the dynamic nature of the fitness landscape, which is especially relevant at the continuously changing host-pathogen interface. Protein variants that are fit in one environment may not be fit in another environment, a concept known as pleiotropy. For example, mutations in S. cerevisiae HSP90 that are beneficial in a high salinity environment are costly in a low salinity environment
.Similarly, many antiviral resistance mutations compromise fitness in the absence of the drug, but enhance fitness in the presence of the drug 56. Different environments, especially variance in temperature, can elicit pleiotropic effects on protein stability. Variants marginally stable at a permissive temperature may not be accessible at elevated temperatures 5, and this pleiotropy
can therefore significantly impact evolutionary trajectories, especially in fluctuating
1.4 Host proteostasis mechanisms assist host protein folding
While proteins fold quickly in aqueous solution, the crowded cellular milieu enhances protein propensity towards aggregation. In cells, these protein aggregates are often toxic, and protein quality control and maintenance of proteome homeostasis (proteostasis) are critical for cellular health and organismal fitness. Thus, organisms are equipped with proteostasis networks, which consist of hundreds of proteins, including protein-folding chaperones and quality control machineries like the ubiquitin-proteasome and autophagy systems 2. Together, these folding
quality control factors work in concert to shepherd nascent proteins to folded, functional conformations while minimizing the
accumulation of misfolded and/or
aggregated proteins ". On a
protein-chaperones folding landscape, the native state is a chaperones
local minimum that can often only be
chaperones
reached by overcoming kinetic
oligomers barriers 2 (Figure 1. 4. 1). Meanwhile,
foldina protein aggregates fall into a global
misfolded intermediate
state minimum that is typically more
~
amorphous
aggregates kinetically accessible. Such
a
native state landscape demands that proteins are
fibrils guided to their native state instead of Figure 1. 4. 1 Protein folding energy landscape. partially folded states or amorphous The protein folding energy landscape is rugged,
where the native state is at a local minimum, but aggregates. terminally misfolded amorphous aggregates and fibrils
are lower in energy. Chaperones promote protein folding by blocking off-target pathways and assisting proteins over kinetic folding barriers. Adapted from Hartl et al.2
Proteino
fanin
ribsm
co-translationalnascent chaperones
begins co-translationally, polypet ide
chain
when chaperones bind to r y HSP7O/4O
terminally
the nascent chain and misfolded
prevent misfolding by
proteasome +co-chaperones
avoiding non-native +ATP I_
contacts (Figure 1. 4. 2) 58. autophagjio Te or I+ATP
Unfolded polypeptides are native state HSP90
system
recognized by exposed chaperonins /+ATP
+ AT P hydrophobic regions, and
bind chaperones such as native state
eukaryotic HSP70/HSP40 Figure 1. 4. 2 Eukaryotic chaperone-assisted protein folding.
for one ATP hydrolysis Representative chaperone pathways that help nascent
polypeptide chains to attain their folded native state and direct cycle, after which they are terminally misfolded proteins for degradation.
released into bulk solution to either continue down a productive protein-folding pathway or
rebind HSP70 for another ATP hydrolysis cycle 59-61. Downstream of the HSP70/HSP40
chaperones, the chaperonins, such as HSP60/110 in eukaryotes and GroEL/ES in bacteria, can enclose select client proteins in a 'cage' that allows proteins to explore a perhaps more limited conformational space, thereby minimizing the entropic costs of folding and preventing aggregation. The HSP90 chaperone system also operates downstream of HSP70/HSP40, by an ATP-driven mechanism that is regulated by several co-chaperones. Generally, chaperones facilitate protein folding by both independent and dependent mechanisms.
ATP-native state or decrease the concentration of unbound non-ATP-native substrates to avoid aggregation 63. Together, these protein-folding macromolecular machines assist protein folding
and prevent misfolding (Figure 1. 4. 1). When proteins are terminally misfolded or form aggregates, the proteostasis network directs them to degradation by the ubiquitin-proteasome or autophagy pathways (Figure 1. 4. 2).
Furthermore, different sub-cellular compartments have distinct but integrated
proteostasis networks. For example, the cytosolic HSP70 and HSP90 systems discussed above have endoplasmic reticulum (ER)-localized counterparts to assist membrane and secretory proteins as they are folded in the secretory pathway 64. These ER protein folding mechanisms
are particularly important at the host-pathogen interface, as they assist the folding of cell surface viral receptors and secreted antibodies. ER-resident proteins often involve complex folding pathways, as they can require glycosylation, formation of disulfide bonds, among other post-translational modifications. Thus, the ER proteostasis machinery contains additional components, such as the protein disulfide isomerase chaperone family, which shuffles disulfide bonds on client proteins, and calnexin and calreticulin, which assist glycoprotein folding and release from the ER 64,65. If ER proteins are terminally misfolded, they can be retro-translocated
to the cytosol, where they are degraded by the ubiquitin-proteasome or autophagy pathways 65. Proper function of cellular proteostasis networks is essential for organismal health. The ability of these networks to fold proteins that would otherwise form aggregates has prompted the hypothesis that protein-folding machinery can impact protein fitness, and thus, protein evolution.
1.5
Host proteostasis both constrains and potentiates host protein evolution
Chaperones are upregulated by cellular stress and can buffer the effects of changing
environmental conditions, such as temperature fluctuation, oxidative stress, or invading
pathogens
64'66.They also buffer the phenotypic effects of genetic change (Figure 1. 5.
1A),
which has been revealed by inhibition of HSP90 in plants, fungi, and Drosophila
4,57-11,13.
For
example, in Arabidopsis thaliana, HSP90 buffers the phenotypic expression of genetic variation,
morphogenic variation in response to environmental changes, and developmental stability
against stochastic processes
5.Similarly, in Drosophila, HSP90 buffers standing morphogenic
variation that is revealed upon disruption of HSP90 activity
1In addition to buffering the
phenotypic effects of standing genetic variation (Figure 1. 5.
1A),
HSP90 has also been
observed to potentiate, or enable, the phenotypic effects of new mutations in budding yeast
(Figure 1. 5.
1B)
12.Although the molecular origins of these results have typically not been
characterized, in principle they may be caused by HSP90 directly engaging an evolving client
protein (termed a primary effect) or mediated indirectly as HSP90 influences the folding of other
endogenous clients that themselves engage relevant evolving proteins (termed a secondary
effect). HSP90-dependent azole resistance in Candida albicans, for instance, is mediated not by
A B
Chaperones as buffers: Chaperones as potentiators:
genetically diverse population genetically diverse population
Chaperone Chaperone
inhibition inhibition
HSP90 directly engaging azole-resistant Erg3 variants, but instead by HSP90-mediated
activation of calcineurin, an HSP90 client that controls responses to various environmental
stimuli 4.
Efforts to look beyond HSP90 and understand how other components of the metazoan
proteostasis machinery modulate endogenous protein evolution (e.g., HSP40/70 chaperones or
protein misfolding stress responses like the heat shock response) have been slowed by a lack
of chemical biology tools to perturb the activities of these systems
26.Recently, work on heat
shock factor 1 (HSF1), the master transcriptional regulator of the heat shock protein chaperones
and cytosolic quality control machinery
67,demonstrated that HSF1 potentiates phenotypic
variation by mechanisms distinct from HSP90
68.Moreover, computational modeling suggests
69,70
that other chaperones, such as the HSP40 family, may have roles in evolution
. In bacteria,
Tawfik and coworkers have shown that the GroEL/ES chaperonin system can alter the
accessibility of endogenous client protein variants. Specifically, GroEL/ES overexpression
potentiates GroEL/ES-dependent enzyme variants containing mutations in the protein core that
were predicted to be more destabilizing than those in GroEL/ES-independent variants
54,71,72.In
another study, the same group found that GroEL/ES-dependent variant expression correlated
with stabilization of folding intermediates, rather than in vitro protein stability
54.Together, these
studies implicate a role for chaperones in shifting the accessible stability range for evolving host
A Basal chaperone B Enhanced chaperone C Reduced chaperone
levels levels levels
folding rate fast
slow
fast
* *0
misfolding 0 * *
slo
--unstable protein inside protein oustide
stability expanded boundary restricted boundary stable can now function can no longer function
Figure 1. 5. 2 Chaperones can shift the boundary of accessible protein stabilities. (A) Protein biophysical boundary model of evolution (see Figure 1. 3. 1). (B) Enhancing chaperone levels can expand the boundary to allow access to additional protein variants. (C) Reducing chaperone levels can contract the boundary and make currently accessible variants inaccessible. In A-C, variants inside the boundary are accessible in the given chaperone environment; variants outside the boundary (in red) are inaccessible. Axes are identical in A-C. Adapted from Powers et al.3
1.6
Viruses engage host proteostasis factors
Identifying the constraints operating on viral protein evolution and adaptation is essential for
preventing and treating viral pandemics. Similar to host proteins, the distribution of mutational
fitness effects for viral proteins can be largely accounted for by considering protein-folding
biophysics
35,73,74.Chaperones and other proteostasis mechanisms are therefore theoretically
well-positioned to address the biophysical challenges created by high mutation rates in viruses.
Though there are rare examples of virus-encoded chaperones
75,
most RNA viruses do
not encode autonomous chaperones or other co-factors to assist protein folding. Instead,
viruses upregulate and engage host chaperones
14-18,76,77(Figure 1. 6. 1), and host chaperone
inhibitors can limit the viability of certain RNA viruses
19-23.For example, HSP90 assists viral
15,16,22polymerase assembly, and HSP90-inhibition can severely reduce replication of influenza
hepatitis B
78,and ebola virus
79,among others. Similarly, HSP70 assists the folding and
assembly of viral proteins
across several viral families
80Beyond promoting folding and
M1
HSPE1CALR CANX
assembly, the heat shock
SP90
1
HSPA2proteins can also regulate
SPA1B
HSPA
SPA8
PDIA6
HSBH1 PDIA
antiviral responses indirectly
NAJA
NNAJ'
through their interactions with PB NAJB NAJB
NAJC7- A
viral
proteins
66'81-83Viral
NAJB1
surface proteins also engage
components of the host's ER
Figure 1. 6. 1 Influenza proteins interact extensively with
host proteostasis factors.
protein folding machinery. The Characterized interactions between influenza
proteins
folding
of
influenza
(colored along perimeter) and human proteostasis factors
(gray ovals)1 4 18 66